
Skin microbiome engineering: Challenges and opportunities in skin diseases treatment
- 28 March 2025
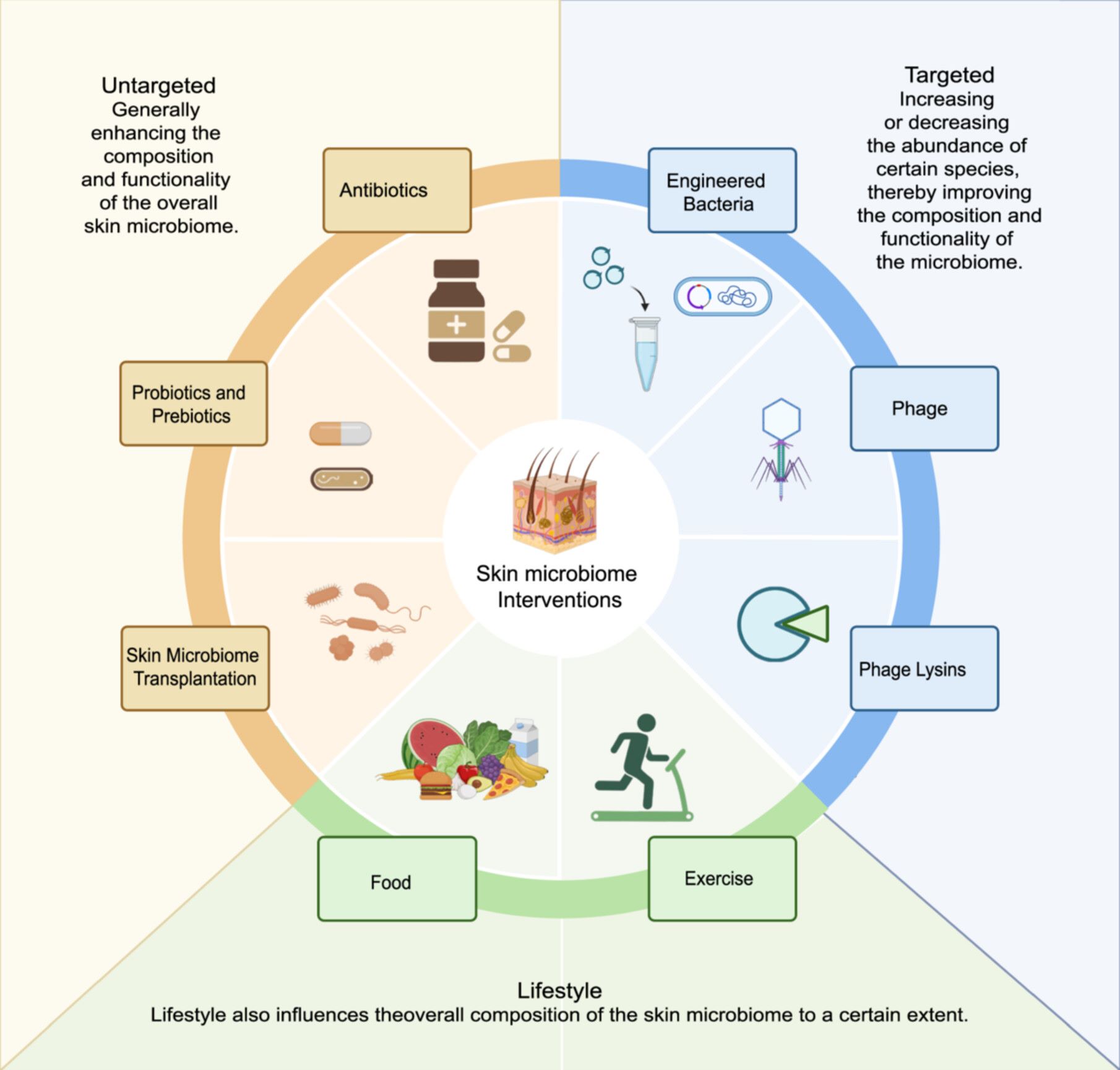
The skin microbiome plays a crucial role in skin health, influencing barrier integrity, immune responses, and disease susceptibility. Various interventions can reshape the microbiome, broadly categorized into targeted and untargeted approaches. Targeted strategies, such as phage therapy, engineered bacteria, and phage lysins, selectively modulate specific microbial populations, restoring balance and mitigating pathogenic influences. Untargeted interventions, including probiotics, prebiotics, skin microbiome transplantation, antibiotics, and lifestyle modifications, aim to enhance overall microbial diversity and stability. These strategies hold promise for innovative dermatological therapies, paving the way for personalized microbiome-based treatments. However, challenges remain in standardization, safety, and long-term ecological impact, necessitating further research for effective clinical translation.
A human skin microbiome reference catalog and the skin microbial landscape of plateau adults
- 11 February 2025
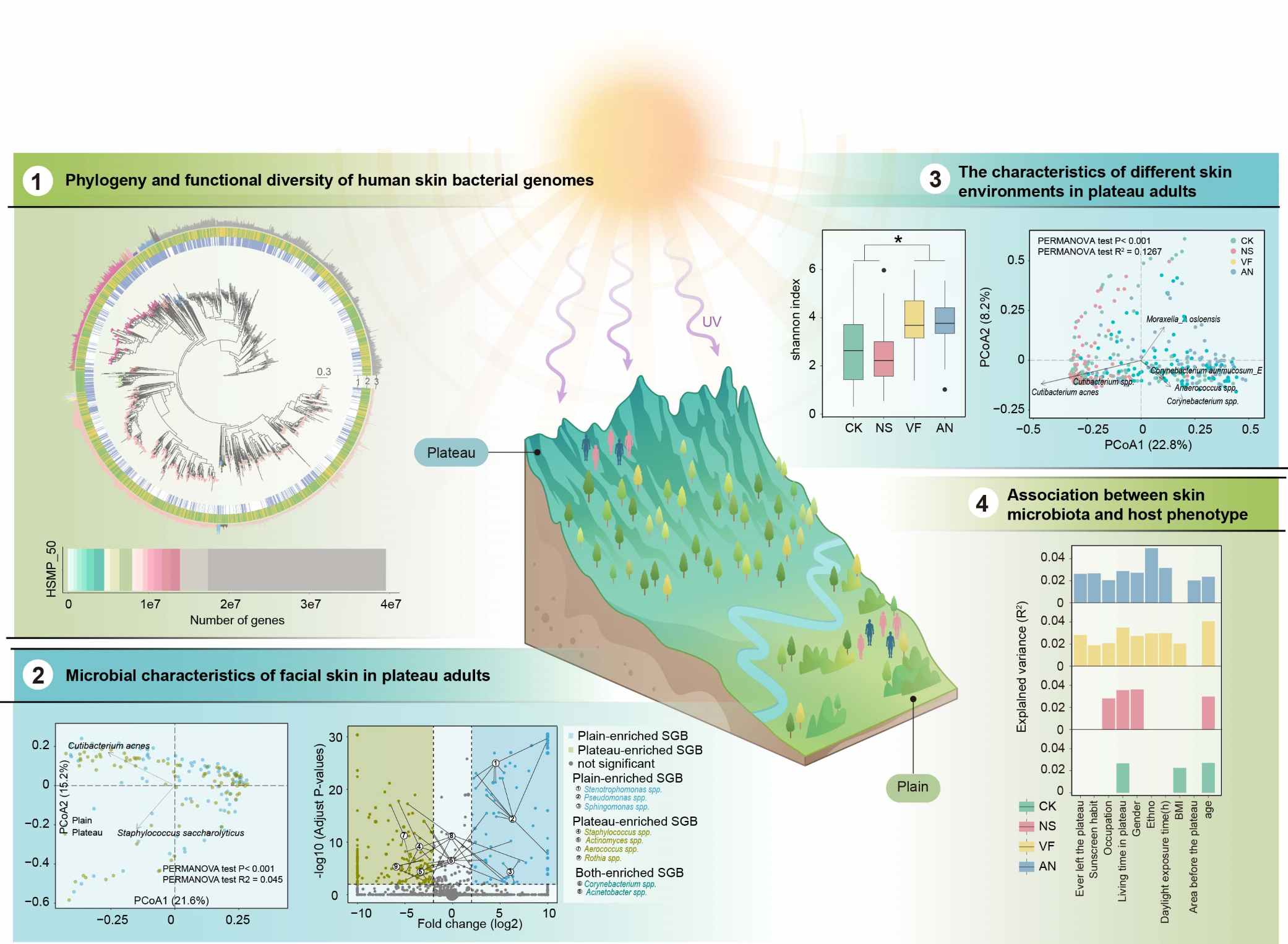
Understanding the skin microbiome is crucial for elucidating its role in health and disease. However, comprehensive studies on the genetic and functional diversity of the skin microbiome in individuals living in extreme environments remain scarce. This work explores the unique characteristics and functional adaptations of the plateau skin microbiome, highlighting its response to environmental stressors, distinct biogeographical patterns, and interactions with host phenotypes.
A novel genotyping system based on site polymorphism on spike gene reveals the evolutionary pathway of porcine epidemic diarrhea virus
- 06 April 2025
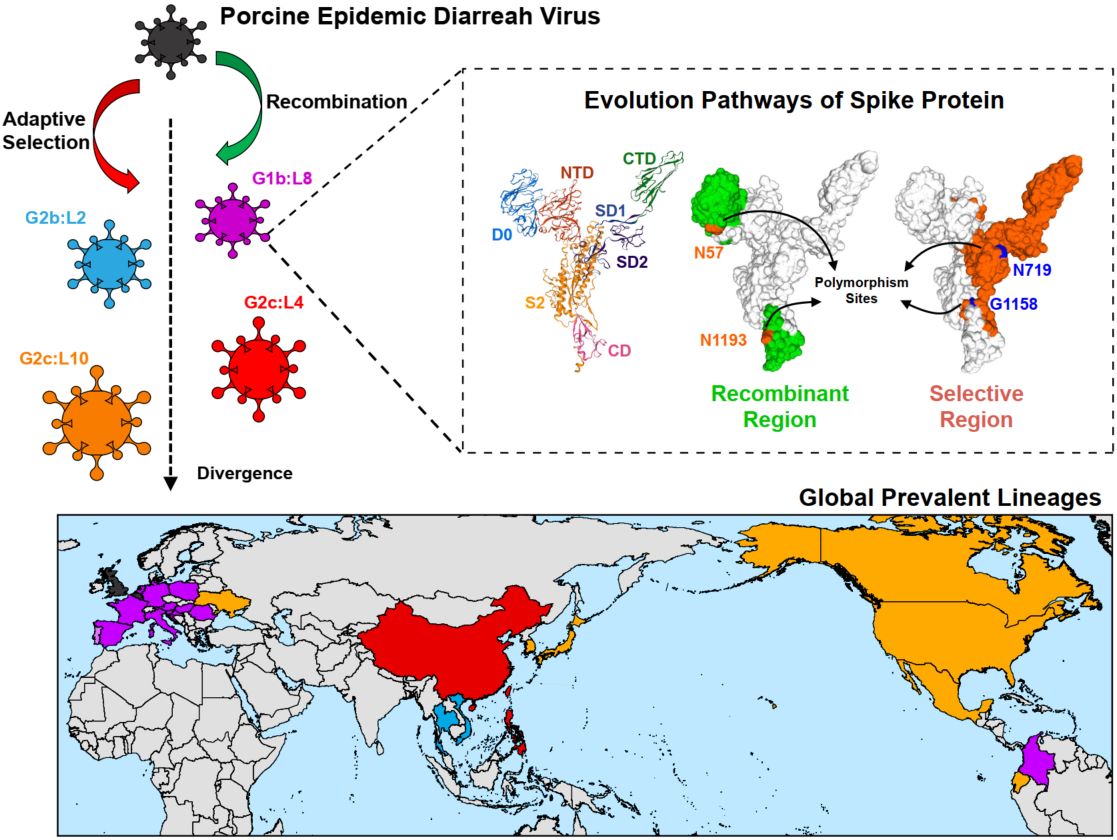
This study focuses on porcine epidemic diarrhea virus (PEDV), a highly virulent and rapidly evolving coronavirus threatening global swine production. We established a novel genotype system of PEDV targeting site-specific polymorphisms in the spike (S) protein. The system demonstrates that frequent recombination events at both termini of the S gene serve as a critical mechanism driving genotypic diversity in PEDV. Together with adaptive evolution, these processes collectively shape the evolutionary dynamics of global PEDV strains. The research provides critical theoretical foundations for understanding PEDV genome evolution patterns and developing prevention and control measures.
Novel machine-learning bioinformatics reveal distinct metabolic alterations for enhanced colorectal cancer diagnosis and monitoring
- 03 March 2025
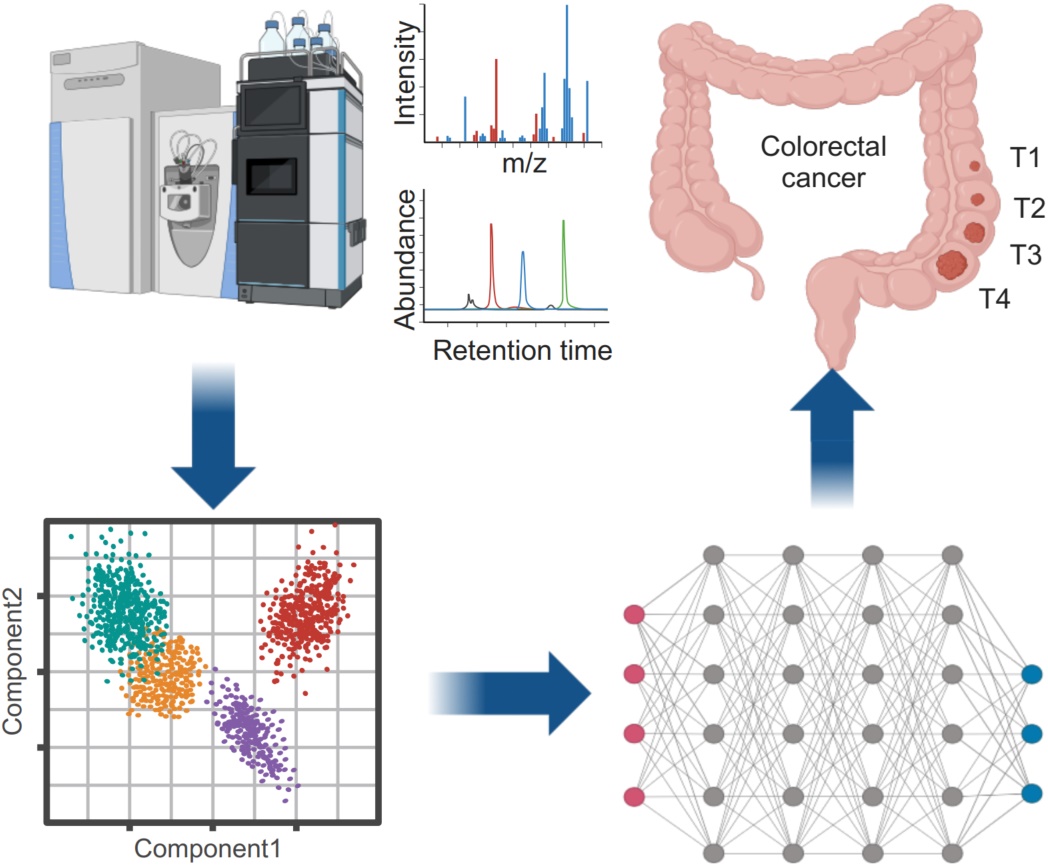
The PANDA pipeline for colorectal cancer (CRC) analysis integrates metabolomic data from LC-MS with machine learning techniques to classify CRC stages and predict biomarkers. Initially, the raw metabolomic data is processed using partial least squares discriminant analysis (PLS-DA) to reduce dimensionality and highlight key features. These selected features are then used to train a neural network, which learns to classify CRC stages (T1–T4) and predict relevant biomarkers. This approach allows for a more accurate diagnosis, early detection, and identification of potential therapeutic targets, contributing to personalized treatment strategies for CRC patients.
Modulation of rhizosphere microbiota by Bacillus subtilis R31 enhances long-term suppression of banana Fusarium wilt
- 18 March 2025

This study indicated that biocontrol bacterium B. subtilis R31 is likely to prevent and control banana Fusarium wilt through regulating the structure and function of banana rhizosphere microorganism flora (especially increasing the microbial abundance of Actinomyces), and help potential biocontrol bacteria grow into the plant roots. There is a convergent secondary succession in microbial communities between naturally suppressive soil and biocontrol R31-treated soil.
Interactions between Parabacteroides goldsteinii CCUG 48944 and diet ameliorate colitis in mice via regulating gut bile acid metabolism
- 18 March 2025
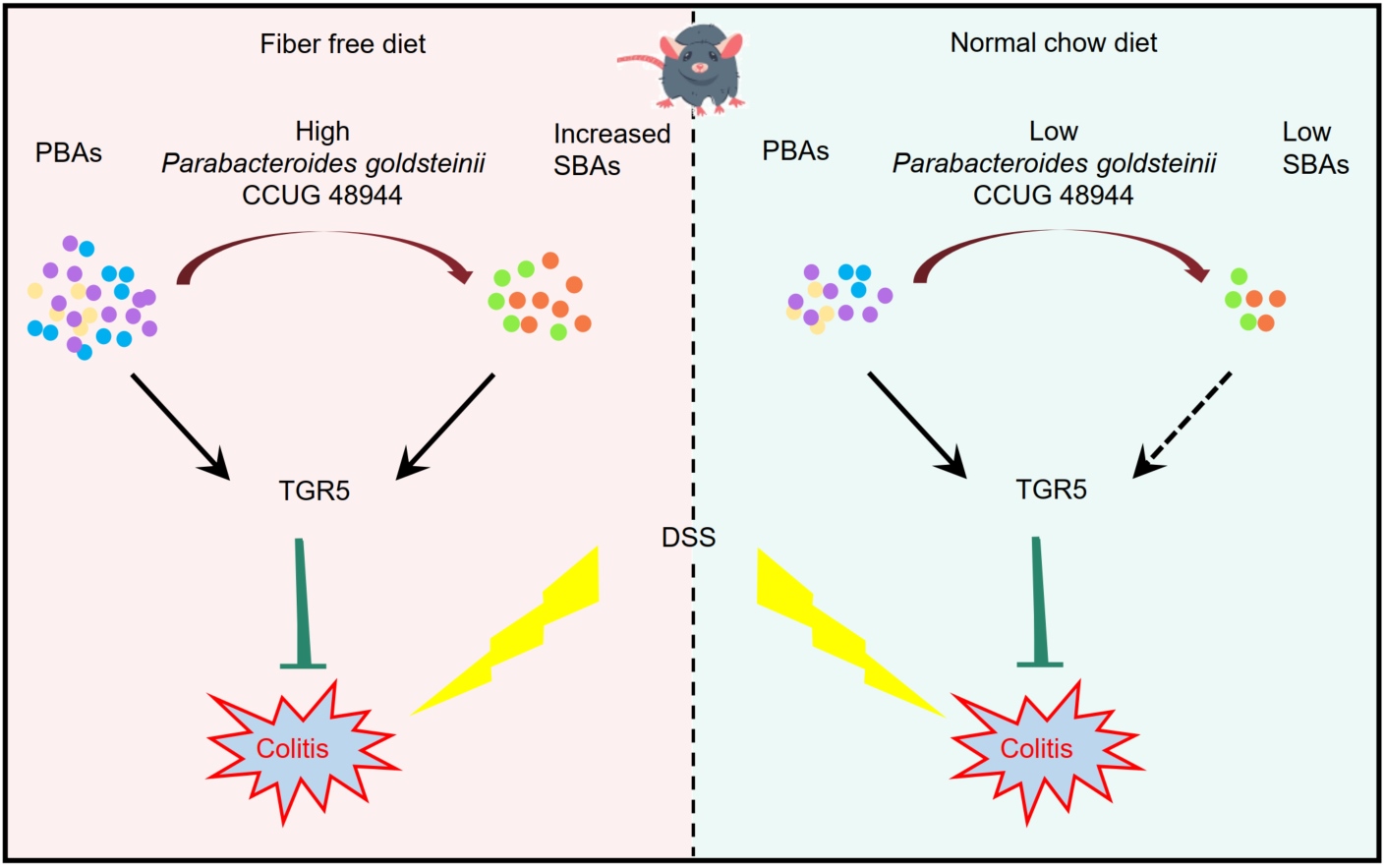
In this study, we explored the therapeutic effects of Parabacteroides goldsteinii (P. goldsteinii) on colitis and its underlying mechanisms. Our results indicate a significant reduction of P. goldsteinii in the stools of inflammatory bowel disease patients. We demonstrate the therapeutic potential of P. goldsteinii in mitigating dextran sulfate sodium-induced colitis and restraining tumorigenesis in the azoxymethane (AOM)/DSS mouse model under a fiber-free diet. Metabolomic profiling further revealed an enrichment of fecal secondary bile acids in response to P. goldsteinii. By employing bile salt hydrolase inhibitors and Takeda G-protein-coupled receptor 5 (Tgr5) knockout mice, we established a connection between the anti-inflammatory effect of P. goldsteinii and fecal bile acids. Furthermore, our findings highlight the modulatory role of diet in enhancing P. goldsteinii's therapeutic potential, underlining the importance of diet in future probiotic research.
Native synthetic microbial communities enhance zha-chili by boosting the fermentation capacity of indigenous microorganisms
- 18 March 2025

Traditional fermented foods in synthetic microbial communities (SynComs). We simulated the production of zha-chili, a traditional fermented food, in the laboratory, intending to address the following questions: How are SynComs constructed, and what is the effect of inoculation with SynComs on the quality of zha-chilli? How does inoculation with SynComs affect microbial community succession?
The performance of different methods in characterizing soil live prokaryotic diversity and abundance is highly variable
- 28 March 2025
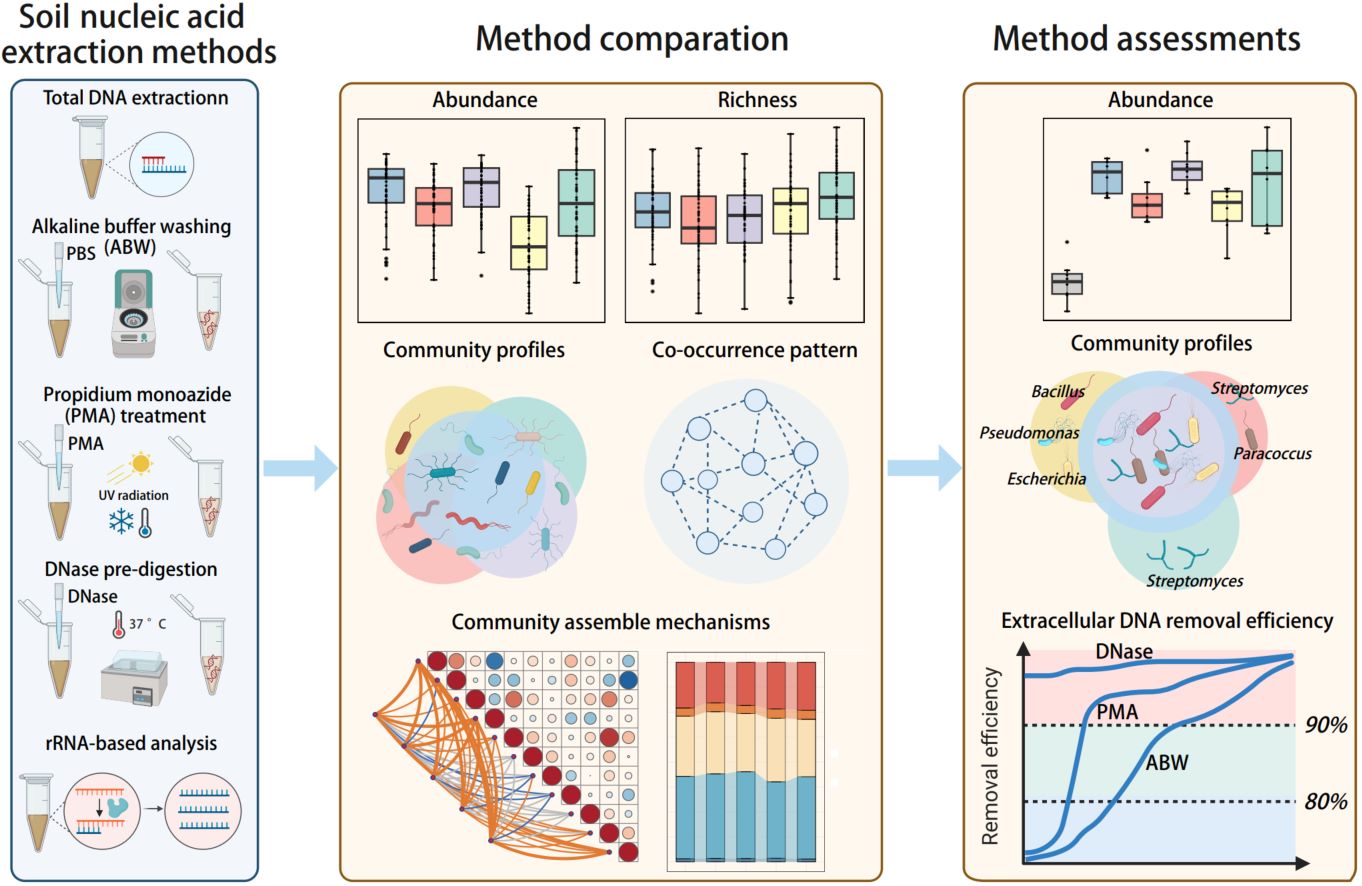
We systematically compared and assessed the commonly used methods for studying live soil microbes, including alkaline buffer washing, propidium monoazide (PMA) treatment, DNase pre-digestion, and rRNA-based analysis, based on soils collected across the western region of China. The results showed that the elimination of extracellular DNA largely affected the analysis of soil prokaryotic abundance, diversity, community profiles, and co-occurrence patterns, but the effects varied considerably across different methods. In contrast, extracellular DNA removal exerted negligible effects on the determination of community assembly mechanisms. We also observed substantial variations in the performance of different methods in characterizing soil live prokaryotic communities, and DNase pre-digestion was recommended for studying soil live prokaryotic communities.
Linking preclinical models to clinical realities: VEGF/VEGFR inhibitors and thrombotic microangiopathy in cancer therapy
- 28 March 2025
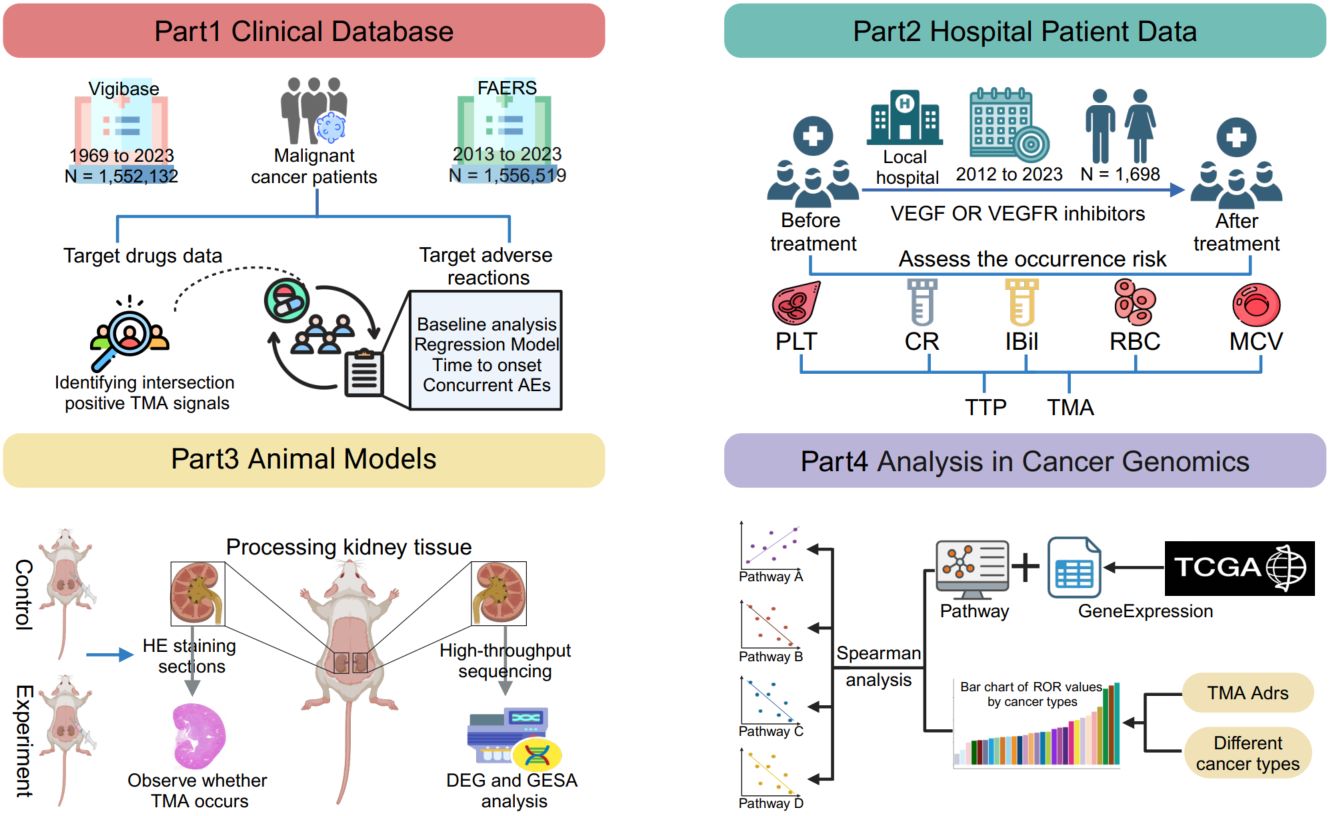
This study investigates the risk of thrombotic microangiopathy (TMA) induced by vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptor (VEGFR) inhibitors in cancer therapy. Using data from the FDA Adverse Event Reporting System (FAERS), the WHO Global Database for Adverse Drug Reactions (Vigibase), and The Cancer Genome Atlas (TCGA), along with clinical analysis of 1,698 patients and animal experiments, it reveals that VEGFR inhibitors cause rapid endothelial damage through complement activation, while VEGF inhibitors trigger delayed TMA via suppressed VEGF signaling. The analysis of TCGA pan-cancer transcriptomic data identified critical pathways involved, such as platelet activation and complement cascade, contributing to TMA development. Animal model studies further confirmed these findings, with hematoxylin and eosin (HE) staining of kidney tissues showing significant endothelial damage, thrombus formation, and mesangial cell proliferation, reinforcing the biological mechanisms. This comprehensive approach underscores the need for regular monitoring of renal function and platelet counts in patients receiving VEGF/VEGFR inhibitors to mitigate TMA risks.
Sex-specific associations of Roseburia with uric acid metabolism and hyperuricemia risk in females
- 01 April 2025
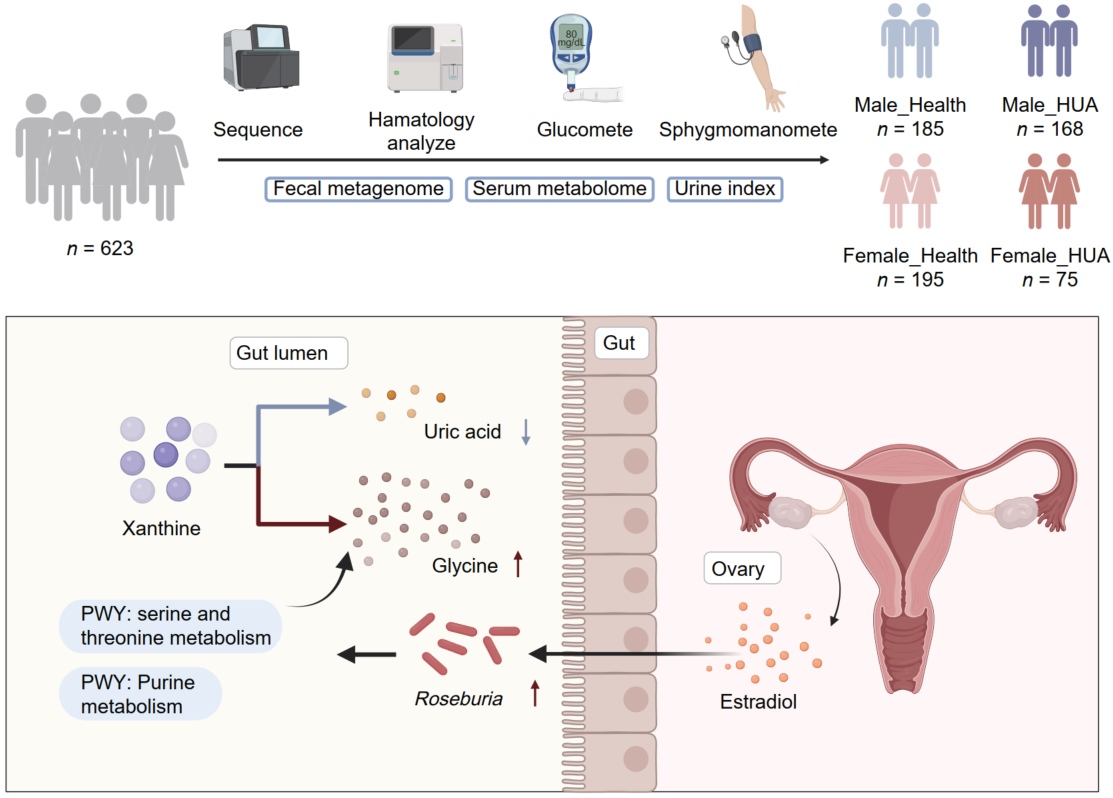
Researchers recruited 623 participants, including 270 females, and used 16S rDNA sequencing to investigate gender differences in hyperuricemia. In healthy females, Roseburia was enriched, positively correlating with estrogen and negatively with uric acid, suggesting it is a potential biomarker affecting uric acid levels in females.
Deciphering comprehensive profiles of pathogenies and resistome of pork using integrating metagenomic and isolation strategies
- 25 February 2025

The pork microbiome was investigated using an integrated approach combining isolation and metagenomic sequencing methods to comprehensively analyze the pathogens and resistome on pork surfaces. The study revealed a large number and diversity of pathogens and resistance genes, potentially originating from air, transportation, water, or cross-contamination. These findings underscore the importance of implementing multifaceted food surveillance strategies to monitor and mitigate these risks effectively.
Selective expansion of gut antibiotic resistome and underlying pathways involved in type 1 diabetes
- 06 March 2025
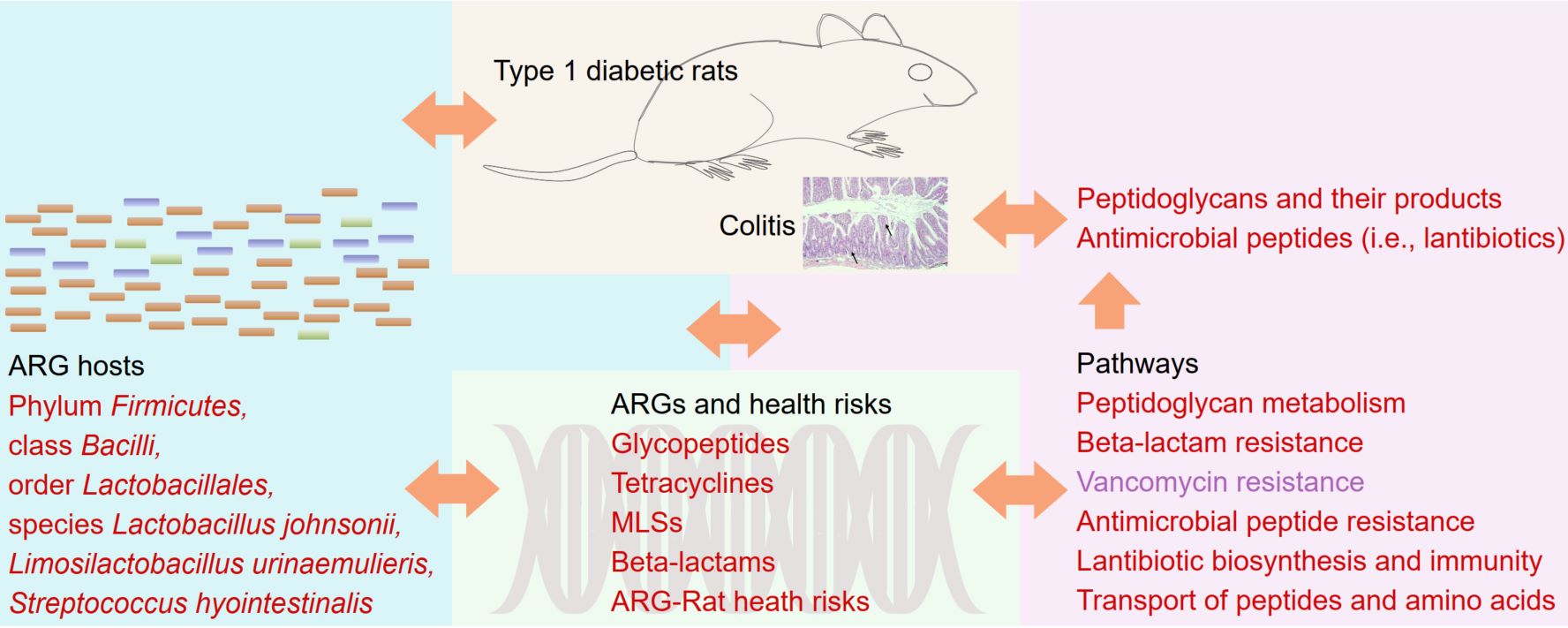
Selective expansion of gut antibiotic resistance genes (including glycopeptides, tetracyclines, macrolide-lincosamide-streptogramins, and beta-lactams) was observed in type 1 diabetic rats. Interestingly, a similar expansion of gut antibiotic resistome occurred in diabetic patients. Selective expansion of gut antibiotic resistome was correlated with hyperglycemia and gut microbial community (particularly order Lactobacillales, such as species Lactobacillus johnsonii, Limosilactobacillus urinaemulieris, and Streptococcus hyointestinalis). Furthermore, activation of peptidoglycan biosynthesis and beta-lactam resistance, disturbances in vancomycin resistance, and activation of antimicrobial peptide resistance, lantibiotic biosynthesis and immunity, and transport of antibiotics, peptides, and amino acids were involved in selective expansion of gut antibiotic resistome in type 1 diabetic rats.
40 years after the discovery of Helicobacter Pylori: Performing optimized “subtraction” for clinical eradication
- 07 April 2025

It has been 40 years since the first discovery of the important human pathogen, Helicobacter pylori (H. pylori). Eradication of the infection continues to pose significant challenges in part because of the global increase in antibiotic resistance. Generally, the “addition” strategy has failed to deliver the desired results for more than a short period and often contributed to the increase in the prevalence of resistant strains. As such, optimization of each component of the treatment strategy has become an increasingly critical focus of research. In this study, we summarized the evolution of H. pylori eradication regimens and the emergence of promising regimens (e.g., high-dose pronto pump inhibitor dual therapy and potassium-competitive acid blocker dual therapy) in clinical practice. Moreover, we conducted an updated meta-analysis incorporating a total of 29 clinical studies regarding potassium-competitive acid blocker dual therapy. Additionally, we also conducted a network meta-analysis regarding the evaluation of first-line regimens for eradicating H. pylori among patients with penicillin allergy. We concluded that the strategy for the first-line treatment of H. pylori infection needs to shift from the traditional “addition” approach to an optimized “subtraction” approach in this era of increasing antibiotic resistance. Dual therapies, particularly those involving vonoprazan and low-dose amoxicillin, have demonstrated both satisfactory eradication rates and patient adherence.
Transfer learning identifies bacterial signatures for cross-regional diagnosis of type 2 diabetes and enable stage-sensitive dietary fiber intervention
- 04 May 2025

DeepMicroFinder is a deep learning framework designed to update the existing disease diagnosis model to generate a transfer model by leveraging region-specific microbiome datasets and transfer learning approach. This framework effectively overcomes the limitation of regional effects in the gut microbiome, enabling accurate cross-regional disease detection. Microbial markers related to type 2 diabetes (T2D) were identified by DeepMicroFinder, and subsequently validated in independent T2D cohorts undergoing dietary fiber interventions.
ViOTUcluster: A high-speed, All-in-one pipeline for viromic analysis of metagenomic data
- 20 May 2025
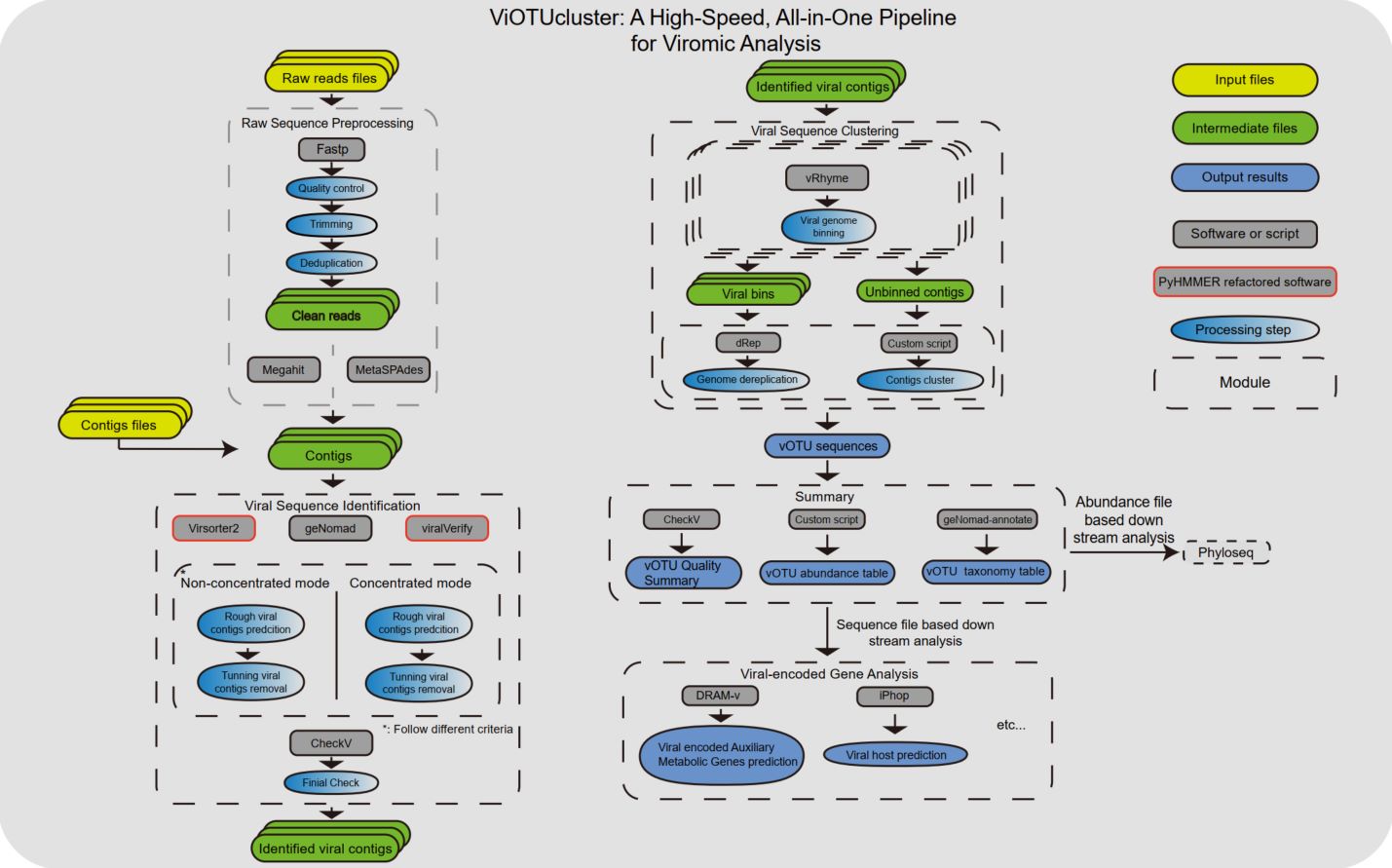
ViOTUcluster is a user-friendly, high-speed, accurate, All-in-one solution that streamlines the entire viromic analysis workflow—from raw reads to the generation of viral operational taxonomic units tables, as well as other key viromic analysis tasks.
Functional and evolutionary characterization of potential auxiliary metabolic genes of the global RNA virome
- 11 February 2025
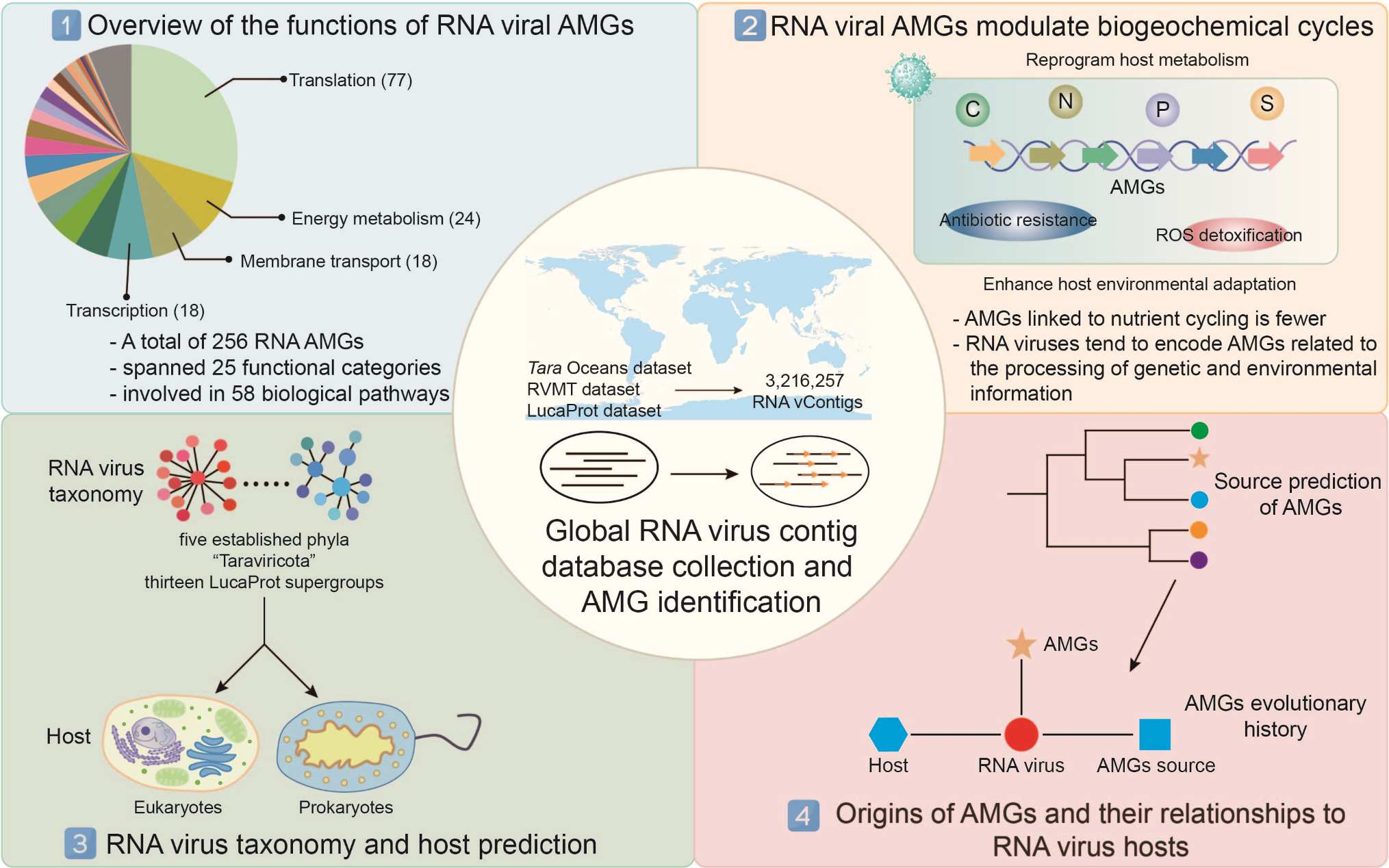
We generated the first comprehensive view of RNA viral auxiliary metabolic genes (AMGs), expanding the known functional type of AMGs by 75%. RNA viruses encode a remarkably high diversity of AMGs, spanning 25 distinct functional categories. Most of these genes are linked to environmental regulation and genetic information processing, with fewer associated with nutrient cycling. Additionally, RNA viruses carrying AMGs are capable of infecting both eukaryotes and prokaryotes, and may acquire AMGs from organisms beyond their predicted host range.
The role of Akkermansia muciniphila in cancer: Mechanisms, therapeutic potential, and challenges
- 27 March 2025
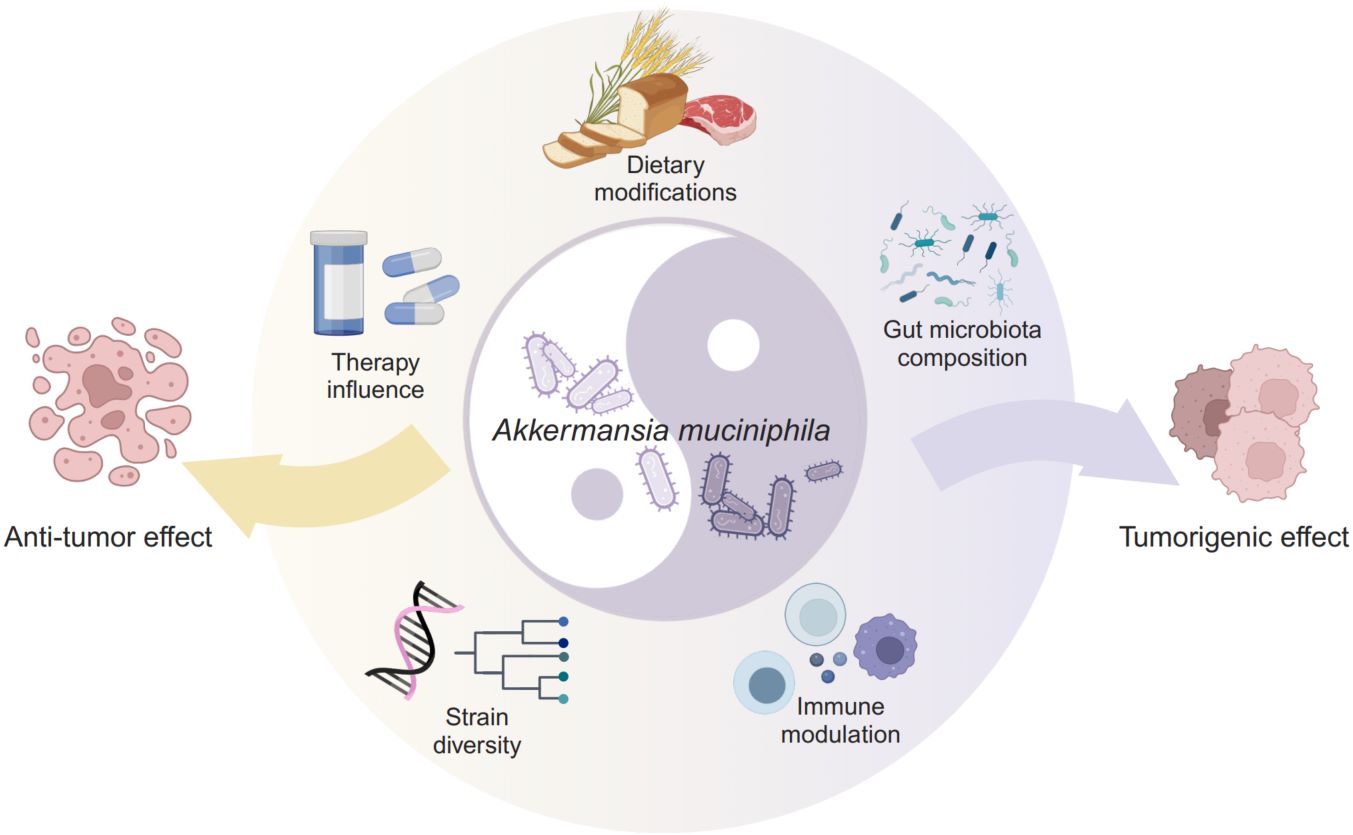
Akkermansia muciniphila (A. muciniphila), regarded as a promising candidate for next-generation probiotic applications, predominantly inhabits the intestinal mucus layer, where it plays a crucial role in maintaining gut barrier integrity and modulating immune responses. Recently, the bacterium has been recognized for its ambivalent influence on cancer, impacting both tumor progression and therapeutic interventions. Research indicates that A. muciniphila might possess both tumorigenic and anticancer capabilities, influenced by factors such as the composition of the gut microbiota, dietary modifications, and immune modulation. There is a compelling need for further studies to uncover the precise mechanisms and optimal use of A. muciniphila in oncology and beyond.
Towards tick virome and emerging tick-borne viruses: Protocols, challenges and perspectives
- 12 May 2025
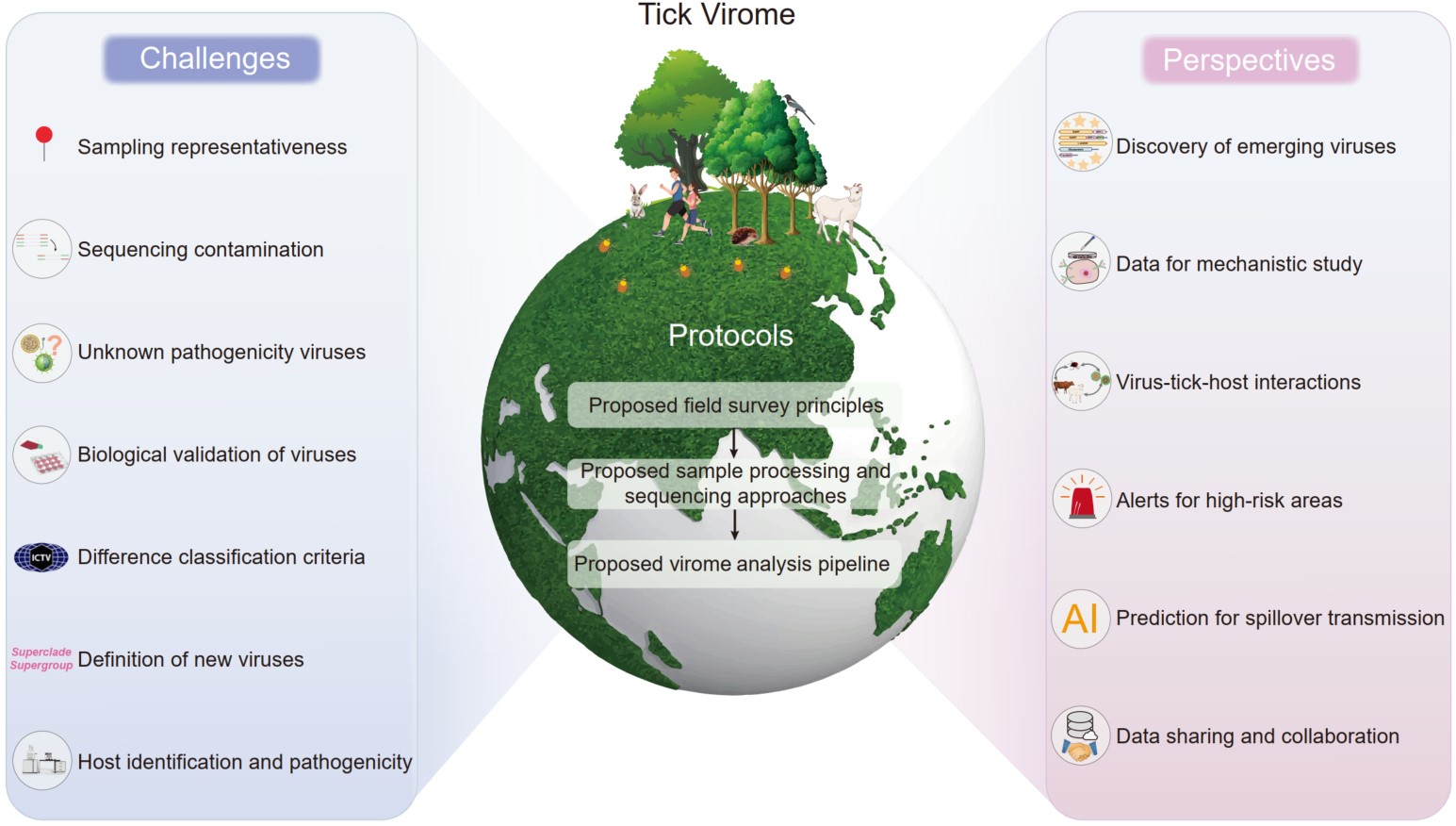
Emerging tick-borne viruses are considered a significant public health threat. The understanding of the tick virome has been revolutionized by high-throughput meta-transcriptomic sequencing, and numerous novel pathogens have been unveiled. However, effective data integration and comparative analyses have been hindered by inconsistent research protocols. In response, standardized protocols are proposed, covering field surveys, sample processing, sequencing library construction, virus assembly, phylogenetic analysis, and classification, offering a unified framework for tick-virome research. Key challenges such as sampling representations, sequencing contamination, and classification discrepancies are also discussed. Furthermore, future perspectives on leveraging bioinformatics and machine learning to trace virus evolution, elucidate virus–tick–host interactions, and enhance surveillance in high-risk areas are outlined in our work. Ultimately, special emphasis is placed on the vital significance of worldwide cooperation and data sharing, which is essential for refining methodologies and guiding targeted interventions to mitigate emerging tick-borne diseases.







