
Harnessing gut microbiota for longevity: Insights into mechanisms and genetic manipulation
- 02 October 2024
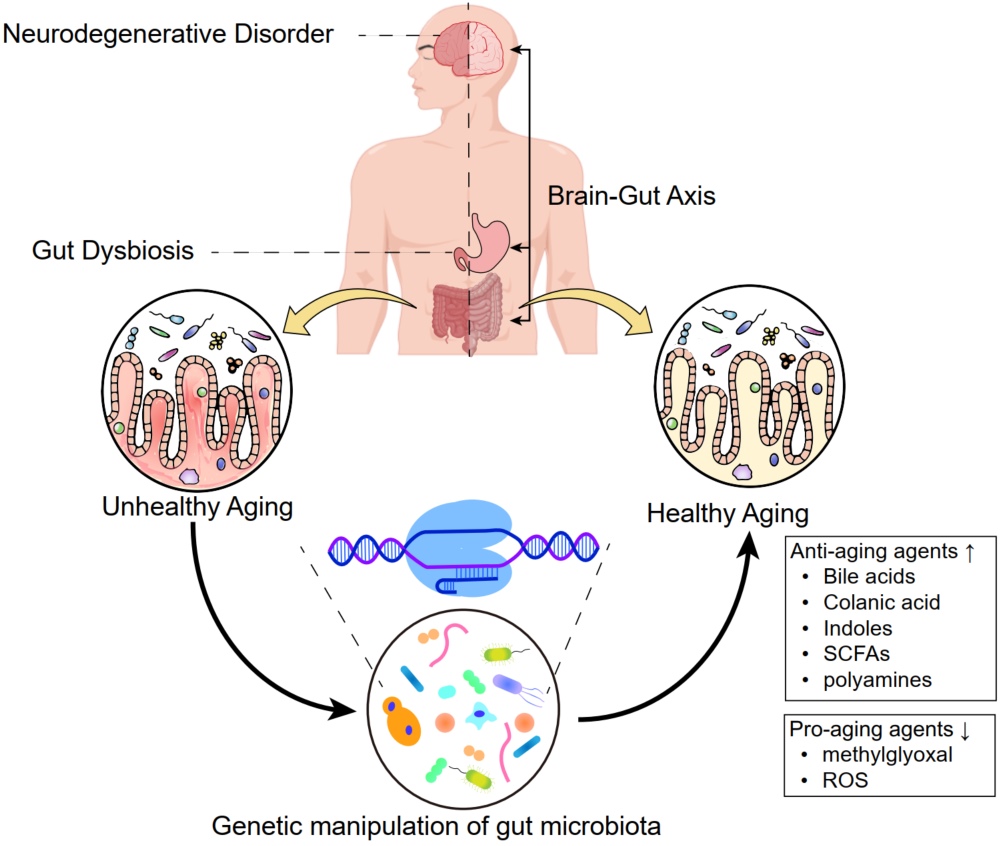
Overview of Gut Microbiota's Role in Aging and Potential for Genetic Manipulation. The relationship between gut microbiota and aging is complex, with significant implications for both health and disease. Advances in genetic manipulation of gut microbiota offer potential interventions to correct dysbiosis, allowing for a microbiome that supports longevity and reduces the risks associated with aging. By targeting specific microbial populations or metabolites, these strategies aim to restore a healthier microbiome, thus promoting healthy aging and mitigating the effects of age-related diseases.
When the microbiome meets One Health principle: Leading to the Holy Grail of biology and contributing to overall well-being and social sustainability
- 10 September 2024
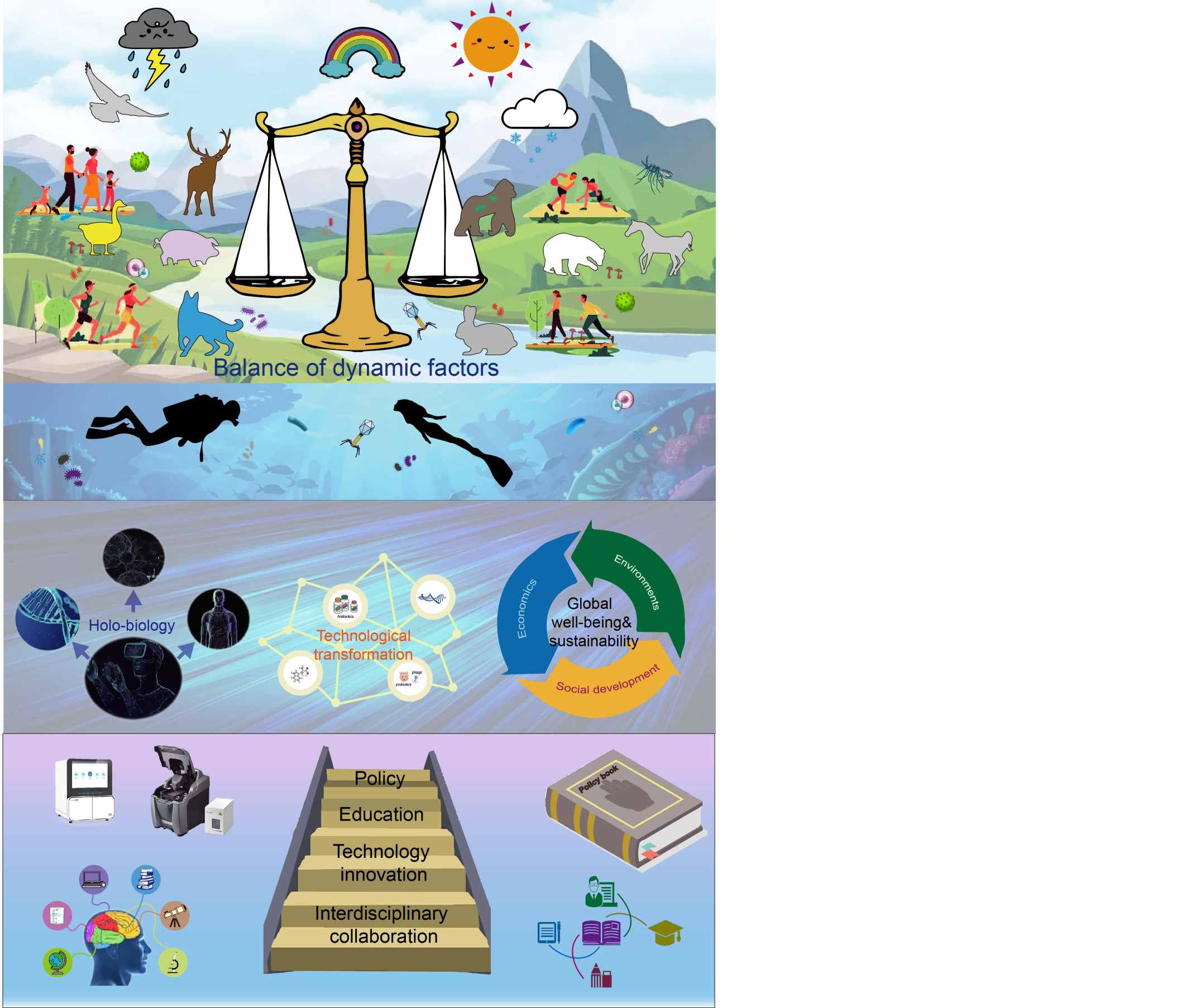
The convergence of microbiome science with the One Health principle heralds a transformative era in biology, prioritizing the collective well-being of humans, animals, and the environment. This review delves into the intricate dance between microbiomes and their hosts, revealing their profound impact on health, nutrient cycles, and climate change. Championing a unified approach to health issues across diverse kingdoms of life, One Health emerges as a holistic strategy, underscored by a proposed universal balance theory, “Balance of Dynamic Factors.” This theory spotlights the equilibrium within microbial and human-animal-environment interactions, offering a revolutionary pathway to global health and social well-being. It paves the way for disease prevention, health equity, and sustainability, all of which are purviews of a balanced ecological system. We navigate the challenges and opportunities of this integrative approach, culminating in a call for action for the incorporation of microbiome science into health policies, precision medicine, legislation, eco-health projects, and education, thereby setting the stage for harmonious coexistence with our planet.
Emerging role of gut microbiota extracellular vesicle in neurodegenerative disorders and insights on their therapeutic management
- 25 October 2024

Gut microbiota extracellular vesicles play (GMEVs) a vital role in maintaining the health of the digestive system and facilitating proper brain functioning. Gut dysbiosis exacerbates gut inflammation, facilitating the entry of harmful substances into the bloodstream. Systemic inflammation has the potential to impair the integrity of the blood-brain barrier. Neuroinflammation and neurodegenerative illnesses can be triggered by the infiltration of inflammatory substances into the brain. A healthy microbiota safeguards the integrity of the intestinal barrier and inhibits the proliferation of harmful pathogens. In addition, GMEVs convey beneficial chemicals and signaling molecules, enhancing intestinal health and reducing systemic inflammation. An intact gut microbiota provides protection against inflammation and supplies essential nutrients and signaling molecules for optimal brain function, therefore playing a crucial role in maintaining general health. Notes: ↑ means increase, ↓ means decrease.
Unveiling microbial communities with EasyAmplicon: A user-centric guide to perform amplicon sequencing data analysis
- 20 November 2024
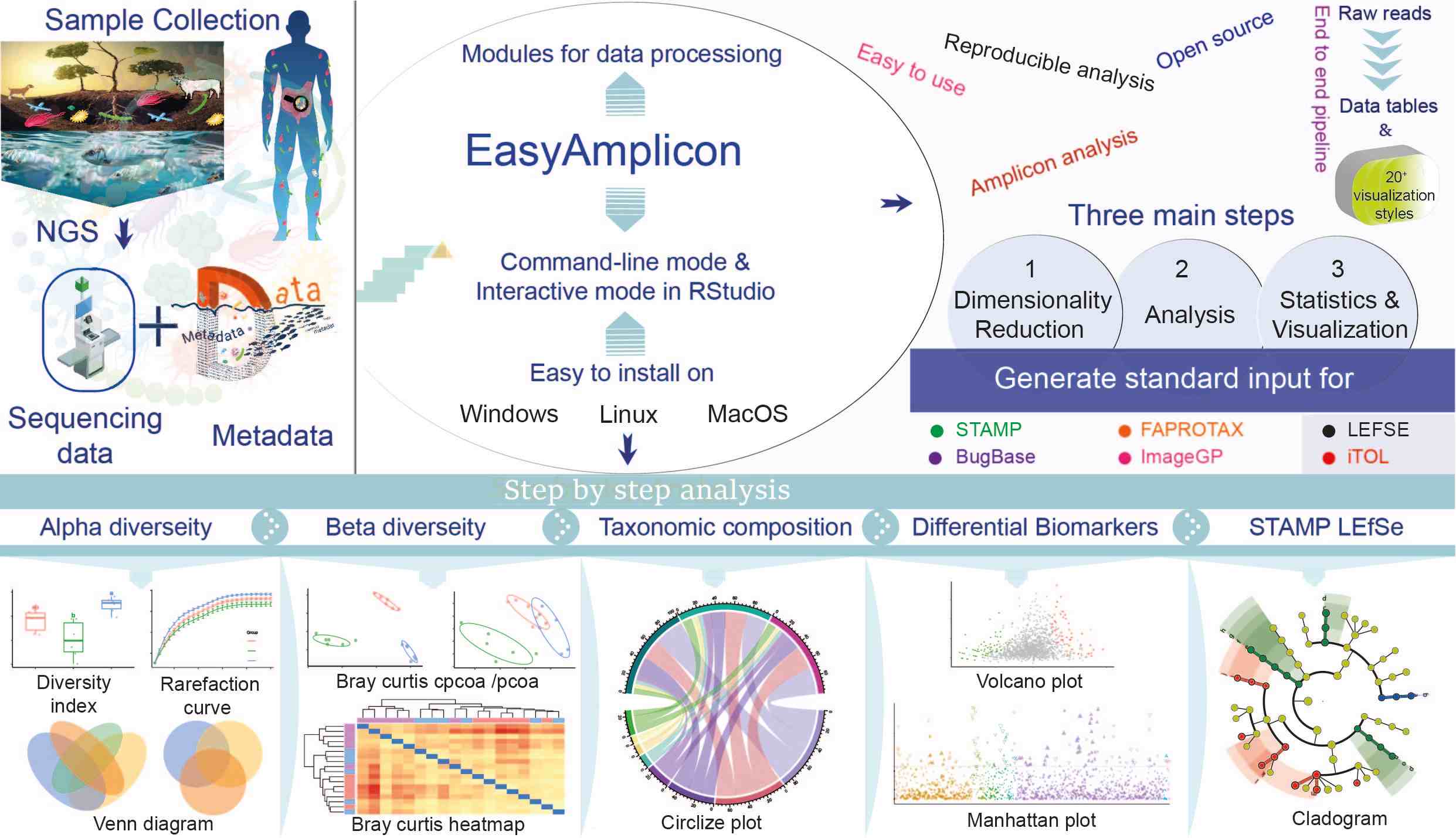
The EasyAmplicon pipeline workflow for amplicon sequencing analysis begins with sample collection and next-generation sequencing, which generates sequencing data alongside crucial metadata for contextual understanding. This user-friendly pipeline is compatible with Windows, Linux, and macOS, offering both command-line and interactive modes in RStudio. The workflow consists of three main steps: dimensionality reduction, analysis, and statistics & visualization, which generate standard inputs for tools like STAMP and LEfSe. Key analyses include α and β diversity, taxonomic composition, and differential biomarkers, emphasizing EasyAmplicon capability to effectively analyzing microbial communities through the integration of sequencing data and metadata.
Culture dependent and independent approaches reveal the role of specific bacteria in human skin aging
- 10 September 2024
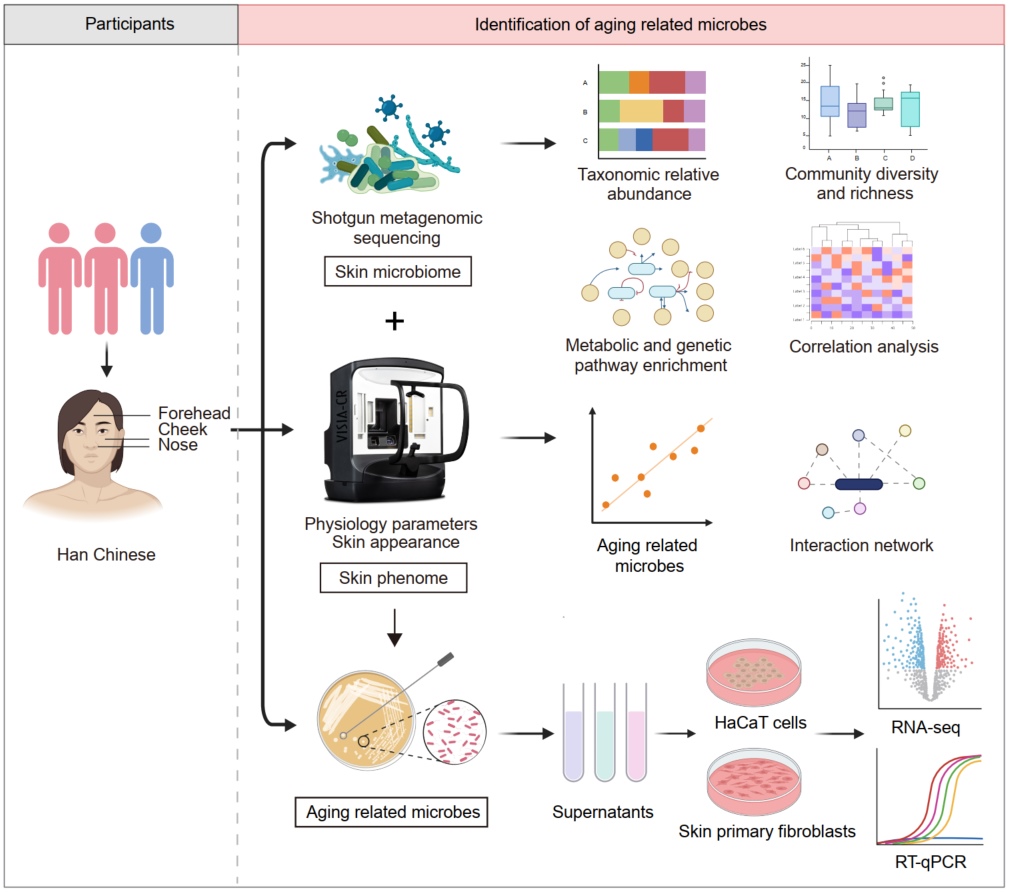
Skin aging is a dynamic process involving a spectrum of phenotypic changes, making it an attractive model for studying microbiome-phenotype interactions. Therefore, 822 facial microbial samples and 14 skin phenotypes from corresponding areas were assessed in a Chinese cohort. Porphyrins and the chronological age exhibited the most significant microbial variability. We further profiled the dynamics of the skin microbiome associated with age and aging phenotypes. Using a multiple linear regression model, we predicted premature/delayed aging-related microbial species, mainly Moraxella osloensis and Cutibacterium acnes. We also validated the biological functions of the host-microbe interactions in vitro. Moraxella osloensis isolated from healthy skin regulates collagen metabolism and extracellular matrix assembly, and promotes cell senescence in human keratinocytes and fibroblasts, making it potentially applicable in the development of antiaging interventions.
Novel insights into immunopathogenesis and crucial biomarkers between primary open-angle glaucoma and systemic lupus erythematosus
- 10 September 2024
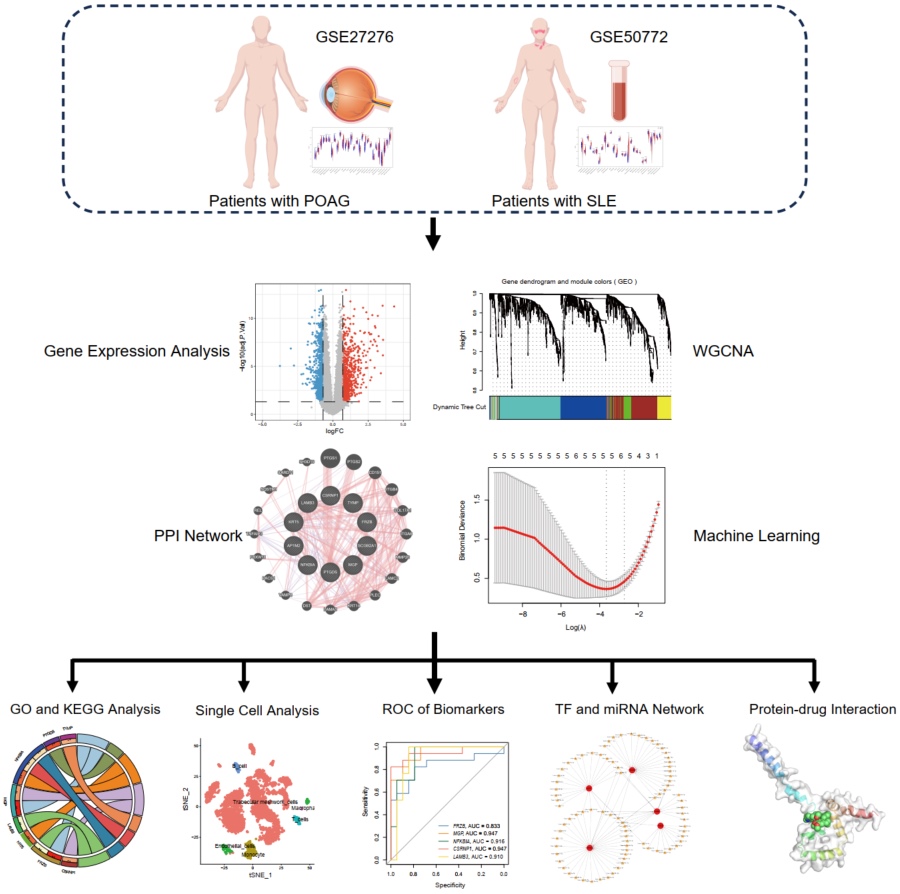
Glaucoma is the primary factor underlying irreversible blindness. Recent studies suggest that the risk of glaucoma significantly increases in patients with systemic lupus erythematosus (SLE); however, the mechanism underlying this association remains unclear. Therefore, this study aimed to identify novel common biomarkers and potential therapeutic drugs for SLE and glaucoma. GSE27276, GSE50772, and GSE148371 datasets were sourced from the gene expression omnibus (GEO) database. An integrated analysis of the datasets for both diseases identified biomarkers and thoroughly examined their biological roles and molecular mechanisms using differential expression analysis (DEA), weighted gene co-expression network analysis (WGCNA), gene enrichment analysis, machine learning, microRNA (miRNA) and transcription factor analyses, immune infiltration analyses, and single-cell transcriptome analysis. Concurrently, molecular docking was used to forecast potential drugs targeting these biomarkers. Finally, reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR) was performed in human trabecular meshwork stem cells to validate the five identified biomarkers. The 10 key genes identified through DEA and WGCNA were predominantly involved in immune, inflammatory, and autophagy pathways. Additionally, machine learning identified five biomarkers, and we established associated transcription factors and miRNA regulatory networks. Immune infiltration analysis indicated an elevated presence of immune cells, including macrophages, T cells, and B cells, in both conditions. Furthermore, the DSigDB database yielded 10 potential therapeutic agents, three of which showed strong binding potential to the biomarkers via molecular docking. The RT-qPCR results confirmed the trend in gene expression. This study uncovered a new link between SLE and primary open-angle glaucoma, identifying biomarkers and mechanisms of immunopathogenesis for future research and treatment strategies.
Integrated omics revealed the altered colonic microenvironment after inhibition of peripheral serotonin synthesis by LP533401
- 14 October 2024
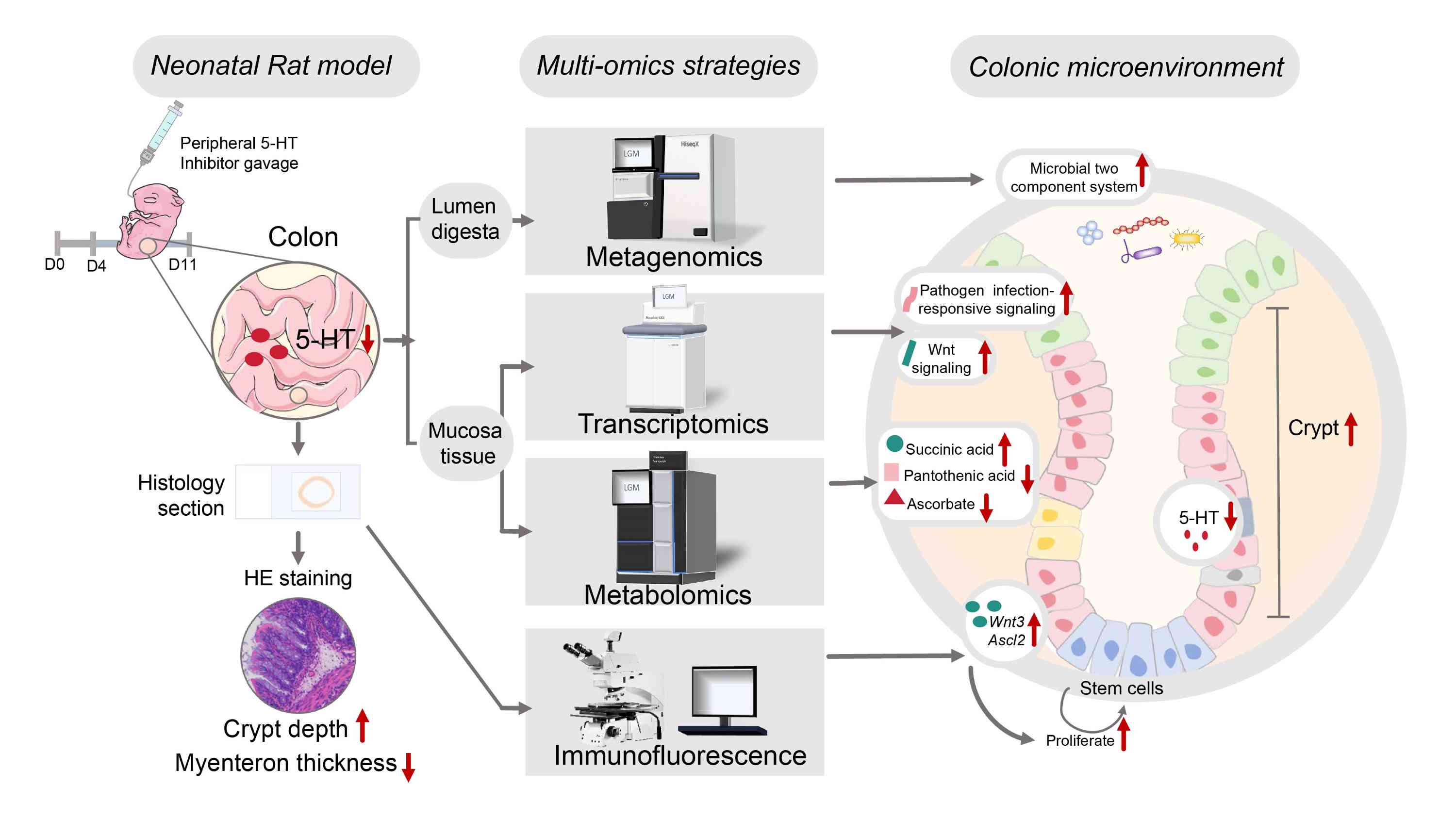
Gut-derived 5-hydroxytryptamine (5-HT), known as serotonin, plays a crucial role in regulating gastrointestinal functions. However, the impact of disruptions in gut-derived 5-HT synthesis on the early gut microbiome and intestinal microenvironment remains unclear. In this study, LP533401, an inhibitor targeting peripheral 5-HT synthesis, was administered orally to neonatal rats starting at 4 days post-birth. By day 11, inhibition of gut-derived 5-HT resulted in altered colonic morphology, characterized by increased crypt depth and reduced myenteric thickness. To investigate the mechanisms underlying these alterations, we employed a combination of metagenomics, mucosal transcriptome, and untargeted metabolomics on colonic samples. Metagenome profiling revealed an upregulation in the microbial two-component system (ko02020) and tyrosine metabolism (ko00350), with minimal effects on taxa abundances. Transcriptome profiling analysis indicated the discriminant expression of genes enriched in pathogen infection-responsive signaling (e.g., Salmonella and Yersinia infection) and the Wnt signaling pathway that affected stem cell proliferation. Consistent with increased crypt depth, marker genes related to cell proliferation were excessively activated. Metabolomics analysis indicated lower ascorbate level and higher succinic acid level, correlating with 5-HT concentrations and increased crypt depth. Additionally, altered metabolic pathways (e.g., nucleotide metabolism, signal transduction, metabolism of cofactors and vitamins) suggested an impact on the colonic function. In summary, early inhibition of gut-derived 5-HT may unfavorably reshape the colonic microenvironment, affecting gut morphology, microbial function, stem cell proliferation, and mucosal metabolism.
Global meta-analysis reveals the drivers of gut microbiome variation across vertebrates
- 04 October 2024
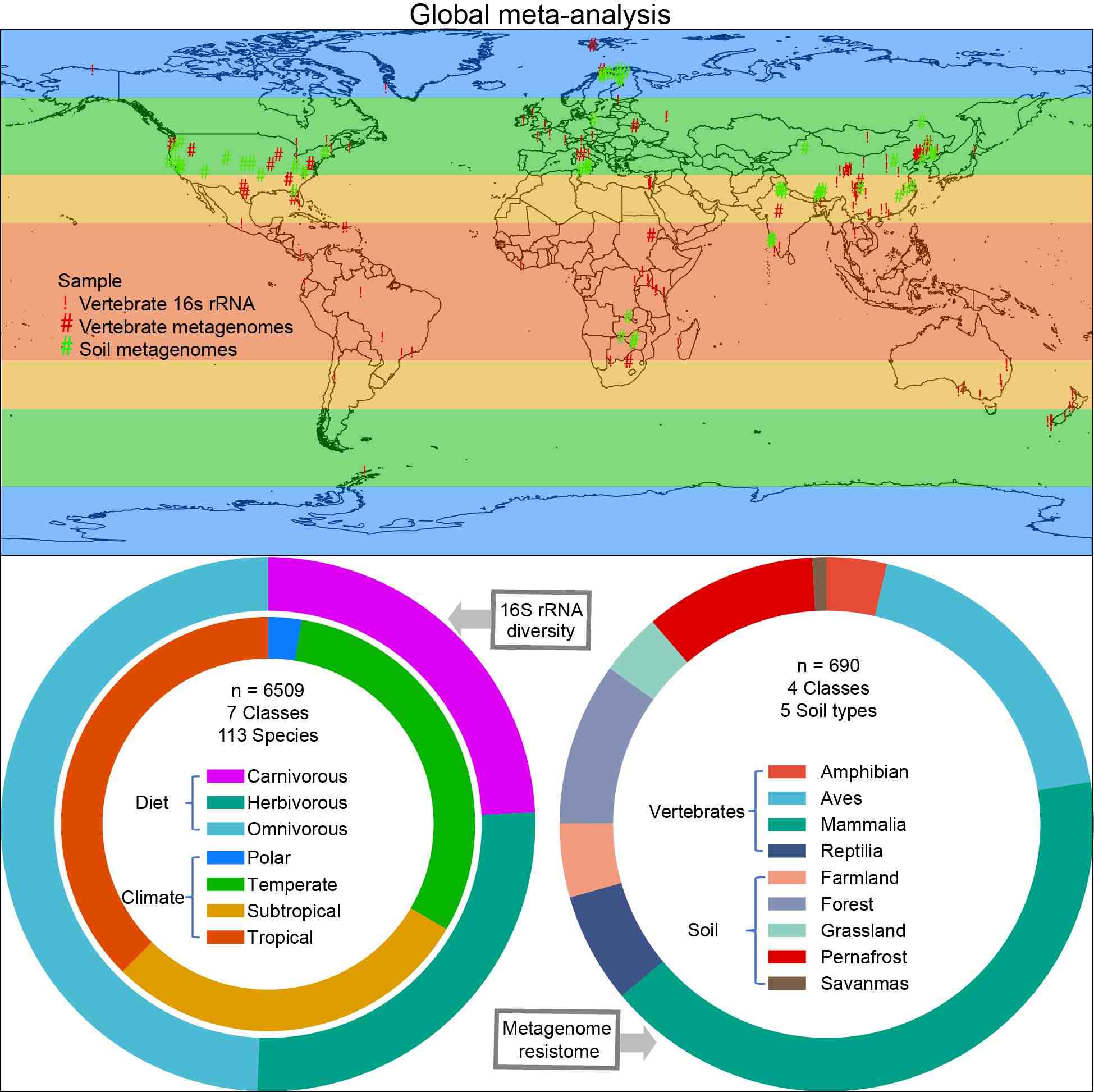
This study analyzed 6508 16S rRNA gene sequence data from 113 vertebrate species worldwide and evaluated the effects of host diet and habitat climate on the diversity of the vertebrate gut microbiome. The results showed that diet and climate factors significantly affected changes in the gut microbiome. At the same time, we also investigated 690 metagenomic samples and found horizontal transfer of antibiotic resistance genes (such as bacA) between the gut microbiome of vertebrates and the sympatric soil environment. This study provides new insights into the diversity and resistance characteristics of the vertebrate gut microbiota and is of great significance for animal protection and antibiotic management.
Multiomics integration unravels genotype-microbiome interactions shaping the conjunctival transcriptome
- 14 October 2024

The effects of genotype and microbiome on molecular phenotypes, such as host gene expression, can vary interdependently. Through an integrative analysis of conjunctival multi-omics data from 120 pairs of twins, we found that incorporating genotype-microbiome interaction terms into gene expression modeling significantly improved prediction accuracy for a substantial proportion of genes, particularly those encoding cell adhesion molecules. We have developed an R package called MicroGenix to screen for potential genotype-microbiome interactions that shape molecular phenotypes using multiomics data.
Genomic adaptation of Clostridium perfringens to human intestine
- 25 October 2024
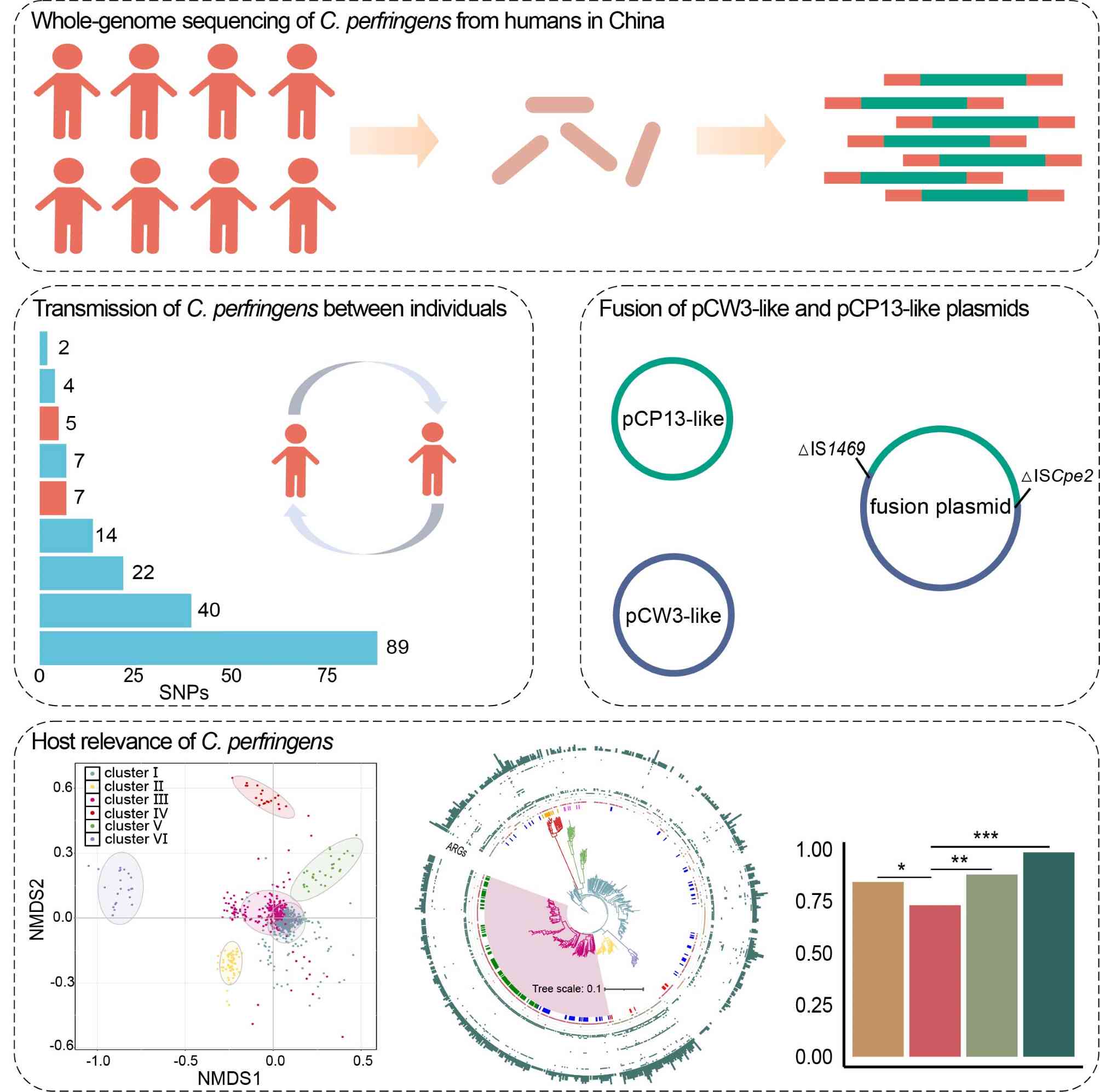
This study performed genomic analysis on Clostridium perfringens from humans in China. There are regional and transregional transmissions of C. perfringens among individuals. cpe was first identified on a pCW3-like and pCP13-like fusion plasmid. Genomic characteristics of C. perfringens are associated with host species.
Dual role of ramR mutation in enhancing immune activation and elevating eravacycline resistance in Klebsiella pneumoniae
- 09 November 2024
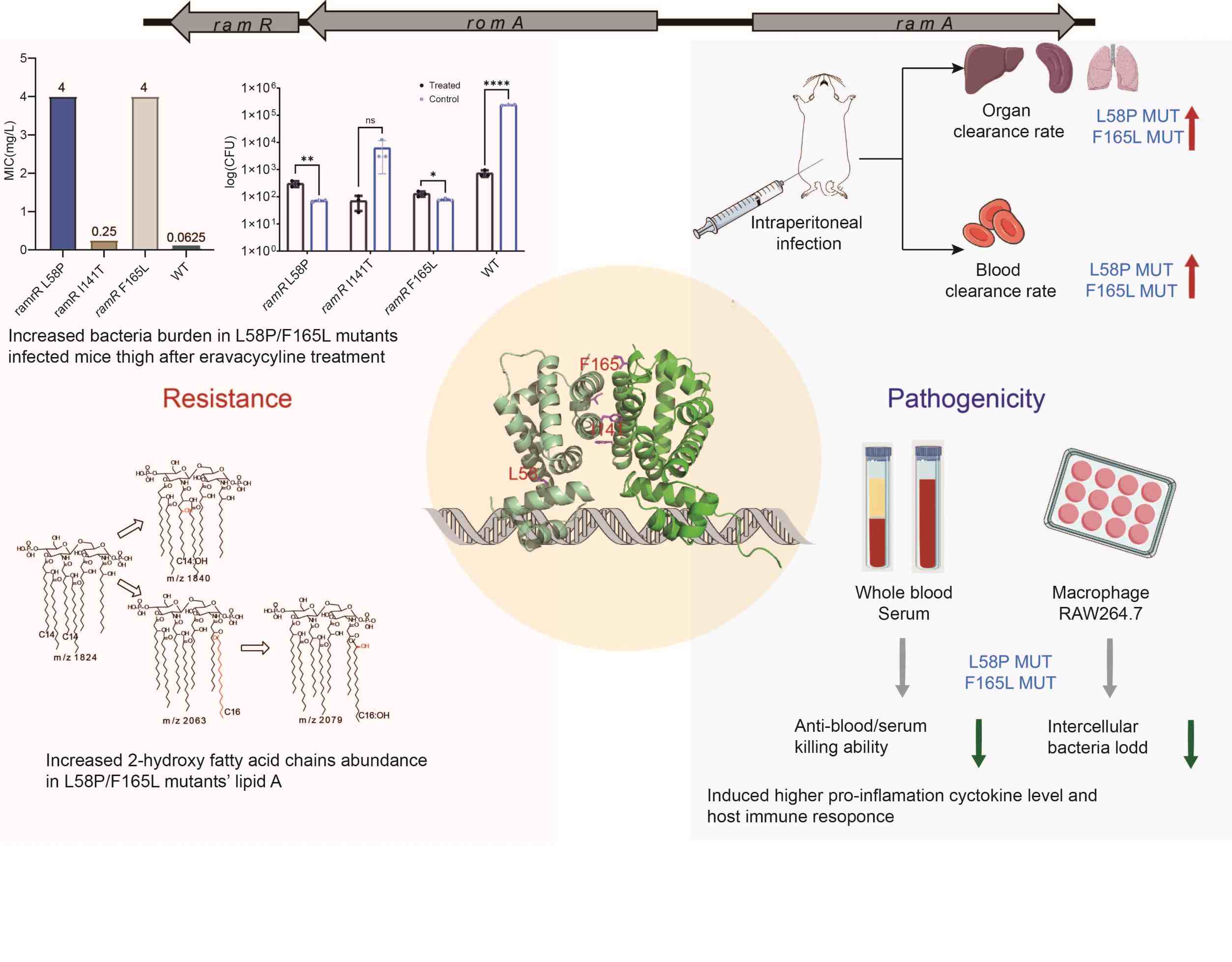
This study performed genomic analysis on Clostridium perfringens from humans in China. There are regional and transregional transmissions of C. perfringens among individuals. cpe was first identified on a pCW3-like and pCP13-like fusion plasmid. Genomic characteristics of C. perfringens are associated with host species.
FoodMicroDB: A microbiome database for composition and time-series research in food
- 01 November 2024
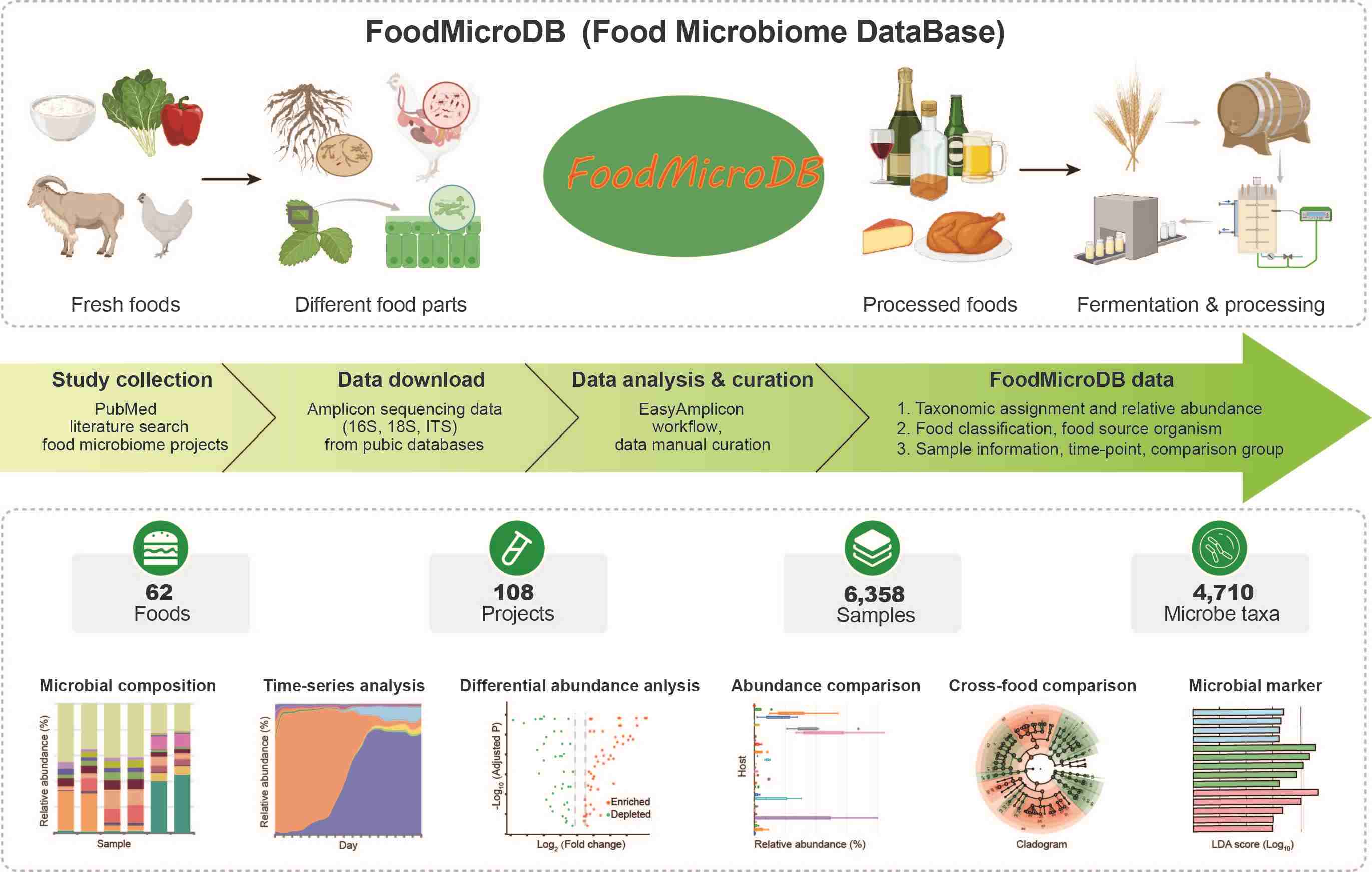
FoodMicroDB is a freely accessible database specialized for food microbiome research, including 6358 amplicon data from 108 meta-taxonomic projects covering 62 foods and 4710 taxa of bacteria, archaea, and fungi. The platform ensures consistent data analysis and metadata curation, facilitating cross-host comparisons. It features versatile data visualization modules, including unique tools for visualizing composition and time-series microbiome data.
Arabidopsis multiomics reveals the role of autophagy in root microbiome assembly
- 05 September 2024
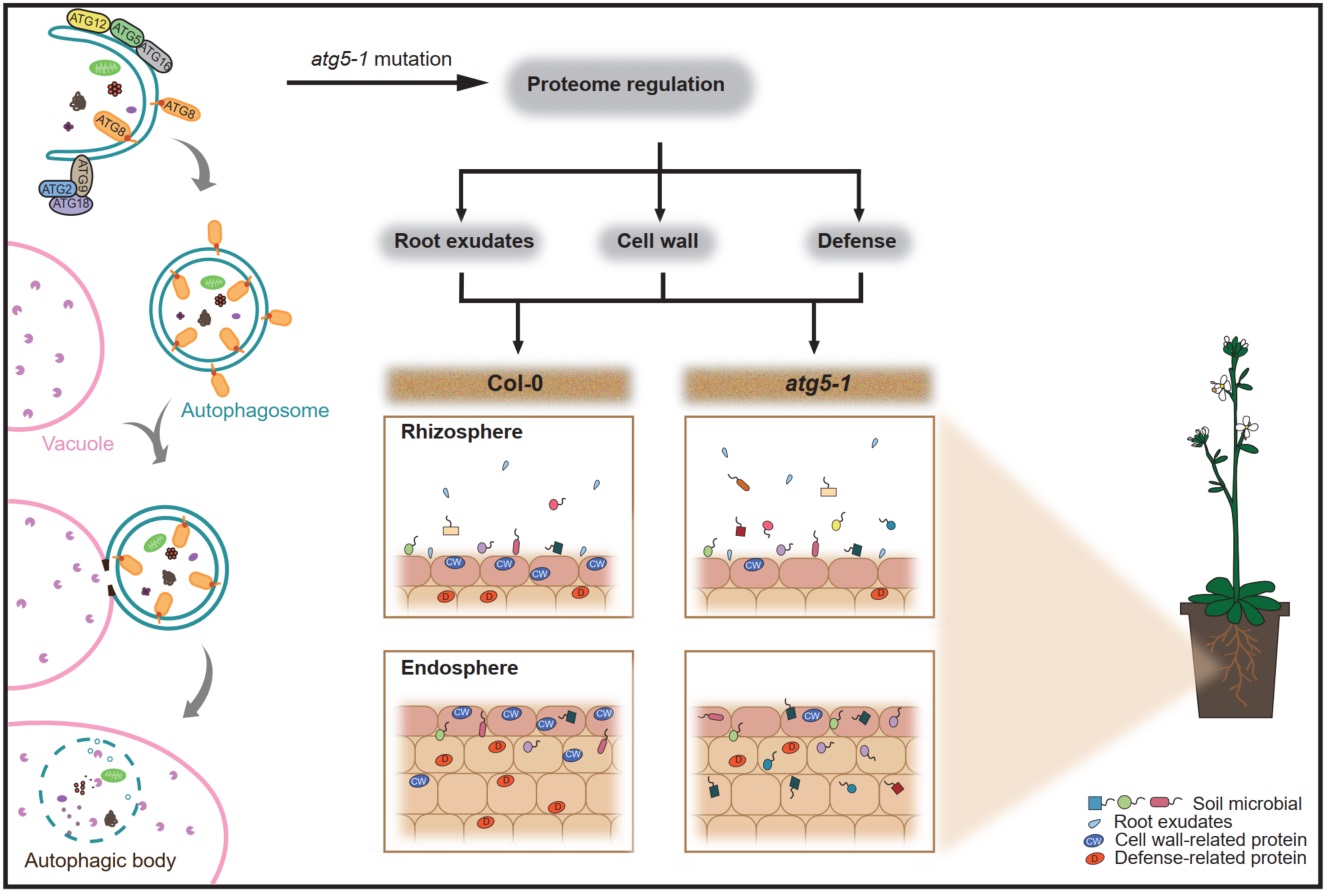
Upon mutation of the core autophagy protein ATG5, disrupted autophagy pathways result in alterations in several biological processes important for plant-root microbe interaction mechanisms, including the expression of cell wall- and defense-related proteins and the secretion of root metabolites, all of which affect the root microbial community diversity.
OUTPOST: A comprehensive analysis software for whole-metagenome shotgun sequencing incorporating group stratification
- 18 September 2024
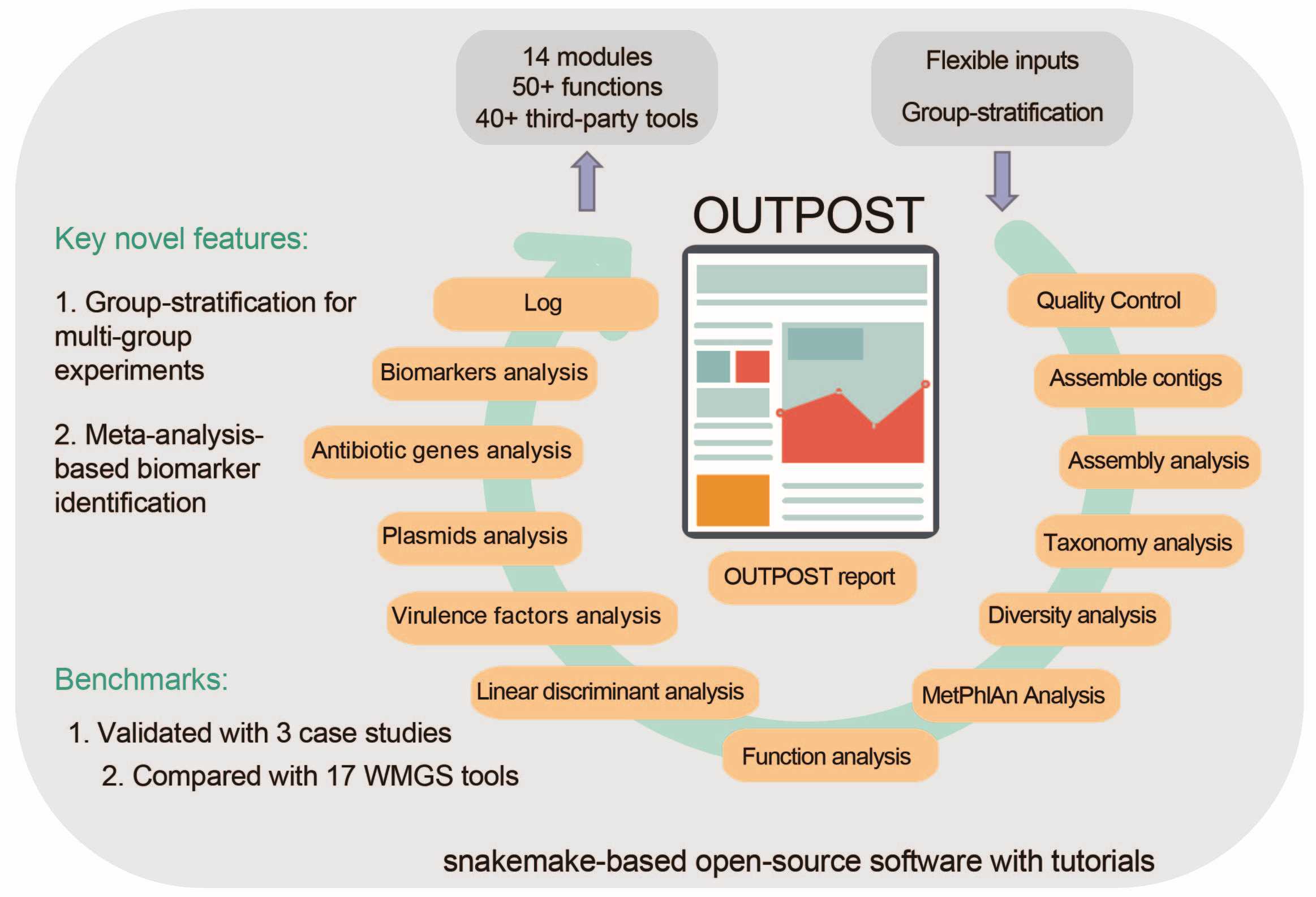
The whole metagenOme shotgun seqUencing sTream Pipeline that is cOmprehensive and uSeful for mulTi groups experiments (OUTPOST) is a comprehensive analysis software for whole-metagenome shotgun sequencing incorporating group stratification, which encompasses 14 modules and boasts over 50 functions, distinguishing itself for its comprehensiveness when compared with 17 existing whole-metagenome shotgun sequencing (WMGS) tools. OUTPOST introduces innovative methods for multi-group experimental designs and meta-analysis-based biomarker identification.
Genome assembly and genomic architecture of a prominent cold-resistant rapeseed germplasm
- 28 September 2024
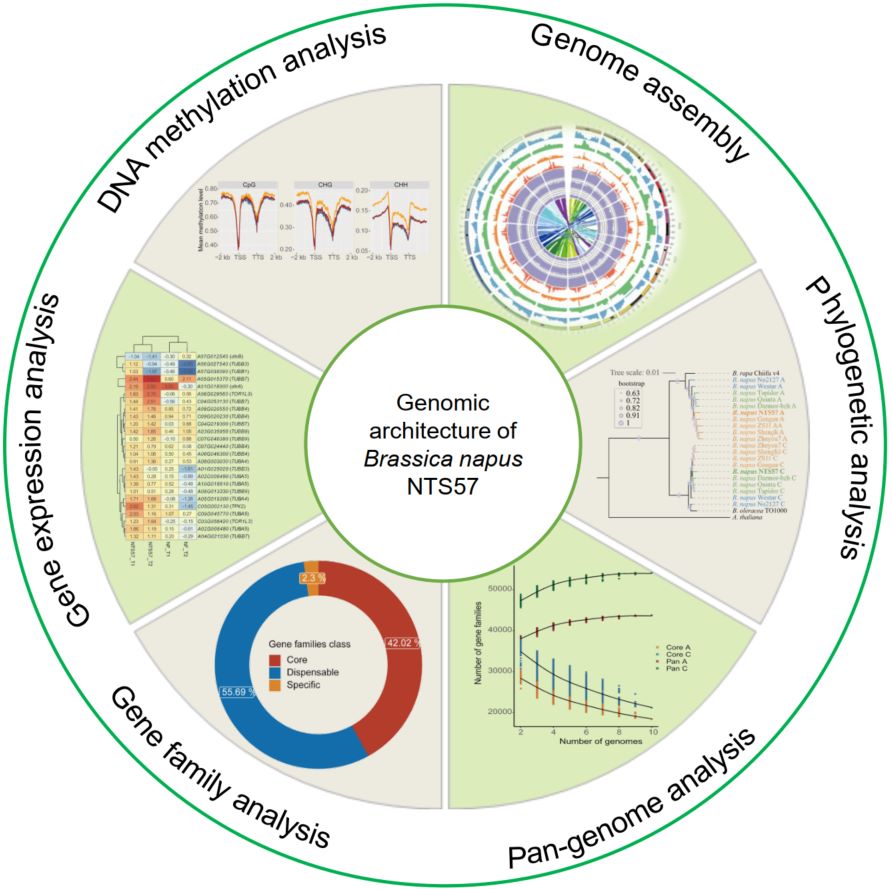
The genomic landscape of cold-tolerant winter rapeseed (Brassica napus, L) has been poorly characterized. We assembled a high-quality reference genome of a prominent cold-tolerant winter rapeseed cultivar, NTS57, and performed phylogenetic and pan-genomic analyses by integrating other reported B. napus accessions. The transcriptome analysis revealed that microtubule-associated biological pathways were much more active in NTS57 under cold stress than in the cold-sensitive variety. Whole genome methylation data analysis revealed that DNA demethylation on protein-coding genes and repetitive elements, especially at CHH sites, is essential for cold response in winter rapeseed.
Pep2TCR: Accurate prediction of CD4 T cell receptor binding specificity through transfer learning and ensemble approach
- 09 November 2024
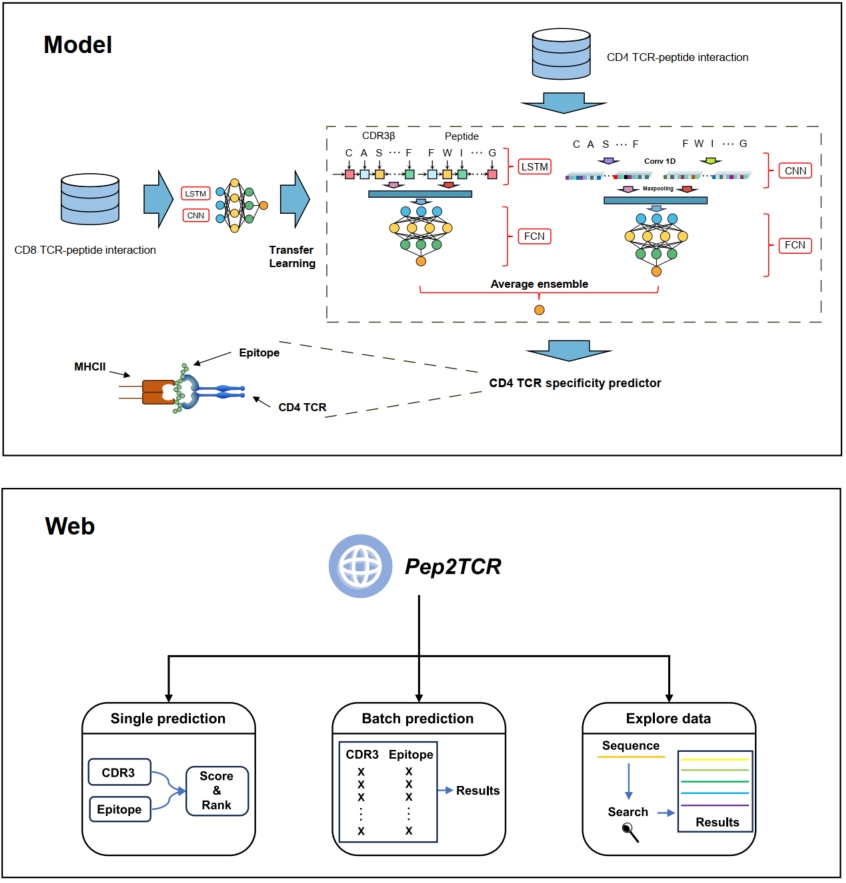
Pep2TCR is an advanced deep learning model designed to predict cluster of differentiation 4 (CD4) T cell receptor (TCR) binding specificity, addressing the challenge posed by limited CD4 TCR data. It shows marked improvement over existing models. Pep2TCR is accessible via a user-friendly website for predicting CD4 TCR specificity at http://pep2tcr.liuxslab.com. This innovative tool holds promise for advancing personalized cancer immunotherapies.
CPStools: A package for analyzing chloroplast genome sequences
- 23 August 2024
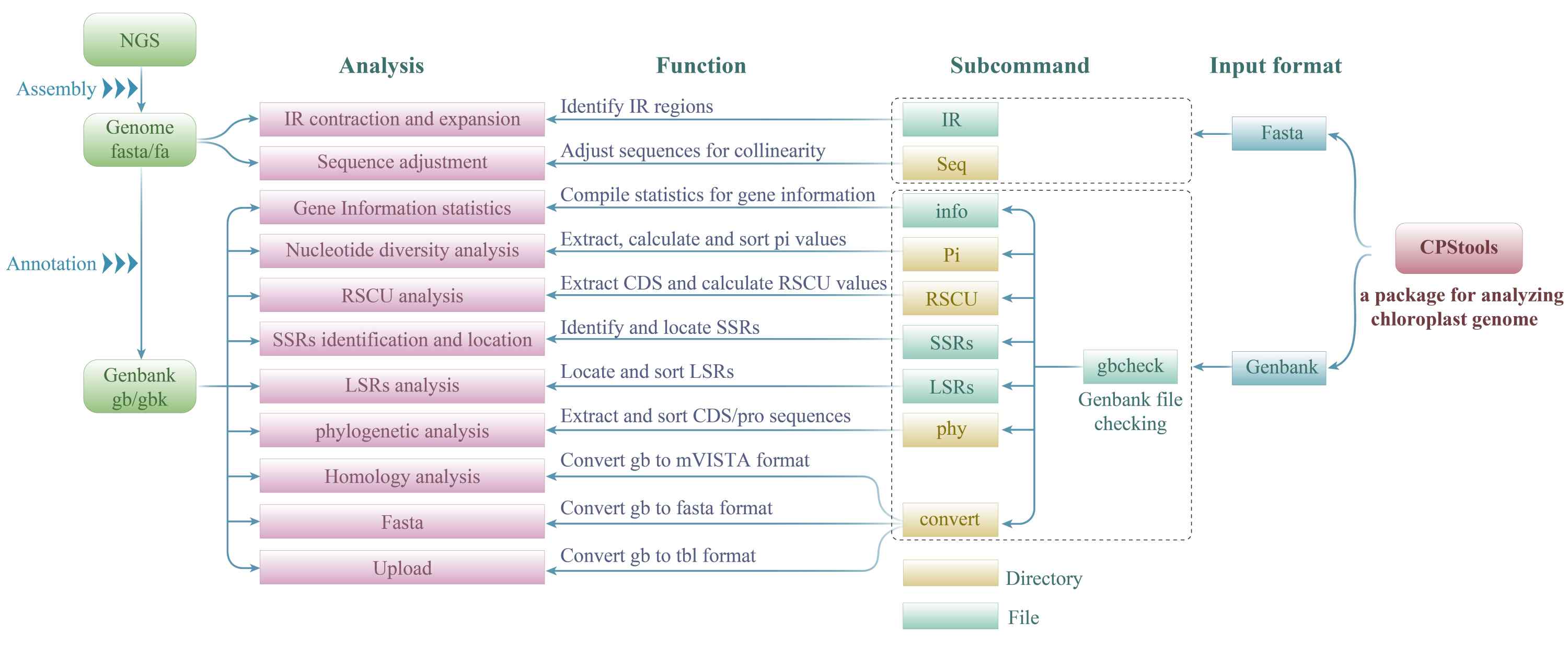
CPStools is a user-friendly software for comprehensive chloroplast genome analysis. It integrates 10 functionalities including Genbank file checking, statistical information generation, sequence adjustment, inverted repeat (IR) regions identification, nucleotide diversity (Pi) analysis, relative synonymous codon usage (RSCU) calculation, simple sequence repeats (SSRs) identification, long sequence repeats (LSRs) statistics, phylogenetic analysis, and format conversion. CPStools handles Genbank or Fasta format inputs, delivering results comparable to other tools while excelling in data preparation for advanced analysis. It uniquely generates consensus merged protein-coding sequence (CDS) or protein sequences from multiple Genbank files, facilitating advanced phylogenetic analysis. CPStools offer reliable results for comprehensive chloroplast genome analysis.
Insights into the identification of antimicrobial peptides: A multidisciplinary observation
- 09 November 2024
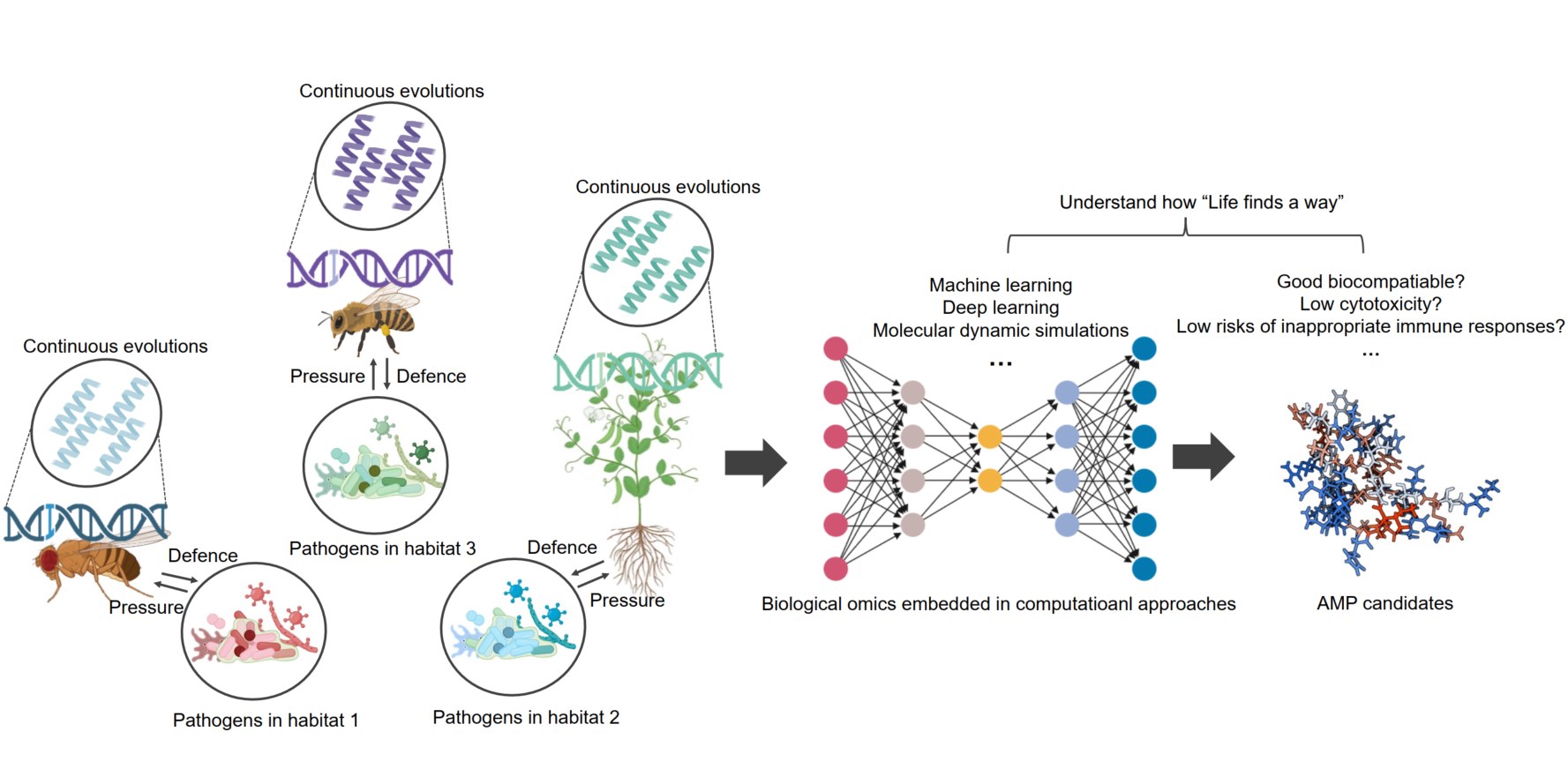
Antimicrobial peptides (AMPs) exemplify the principle of “Life finds a way” by adapting to diverse environmental niches. These peptides exhibit remarkable specificity, targeting pathogens unique to their respective habitats. Understanding the ecological context of AMPs not only clarifies how these peptides selectively target pathogens while remaining nontoxic to hosts, but also underscores the potential of computational approaches in AMP discovery and design. By integrating biological omics data with computational modeling, researchers can develop novel AMPs tailored to combat specific microbial threats, paving the way for innovative therapeutic solutions in an ever-changing microbial landscape.







