










From prediction to actionable mechanisms: Explainable multi‑omics AI for farm‑to‑fork postharvest preservation
- 09 June 2026
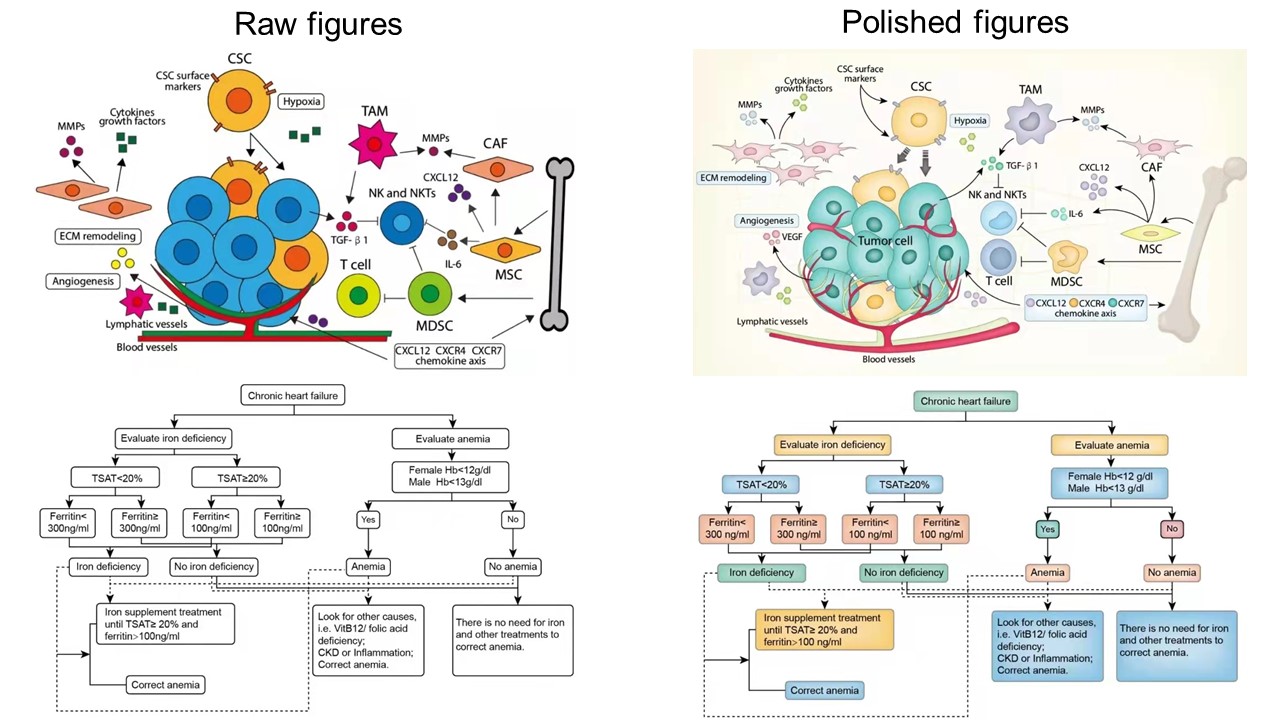
Graphical overview of explainable artificial intelligence (XAI) for farm-to-fork postharvest preservation. Postharvest deterioration accumulates across orchard, packhouse, refrigerated transportation, warehouse, and distribution stages under fluctuating temperature, humidity, atmosphere, and mechanical stress. Multimodal data streams, including host omics, microbiome profiles, environmental sensing, RGB/hyperspectral/thermal imaging, spectroscopy, key genes, and logistics records, are integrated through a data lakehouse and analyzed by postharvest XAI models. Explainable modules, including SHapley Additive exPlanations (SHAP)/local attribution, graph neural network (GNN) explanation, pathway-constrained models, counterfactual reasoning, and stability/faithfulness auditing, convert black-box spoilage-risk prediction into interpretable biological mechanisms. These mechanisms guide actionable interventions such as antioxidant coating, elicitor spray, biocontrol consortia, packaging optimization, and gene-targeted strategies. Validation through storage trials, sensory evaluation, microbial testing, and sequencing closes the loop from prediction to explanation, intervention, and validated decision support.
A spatiotemporal single-cell atlas of porcine development reveals regulatory dynamics and cellular targets of domestication
- 04 June 2026
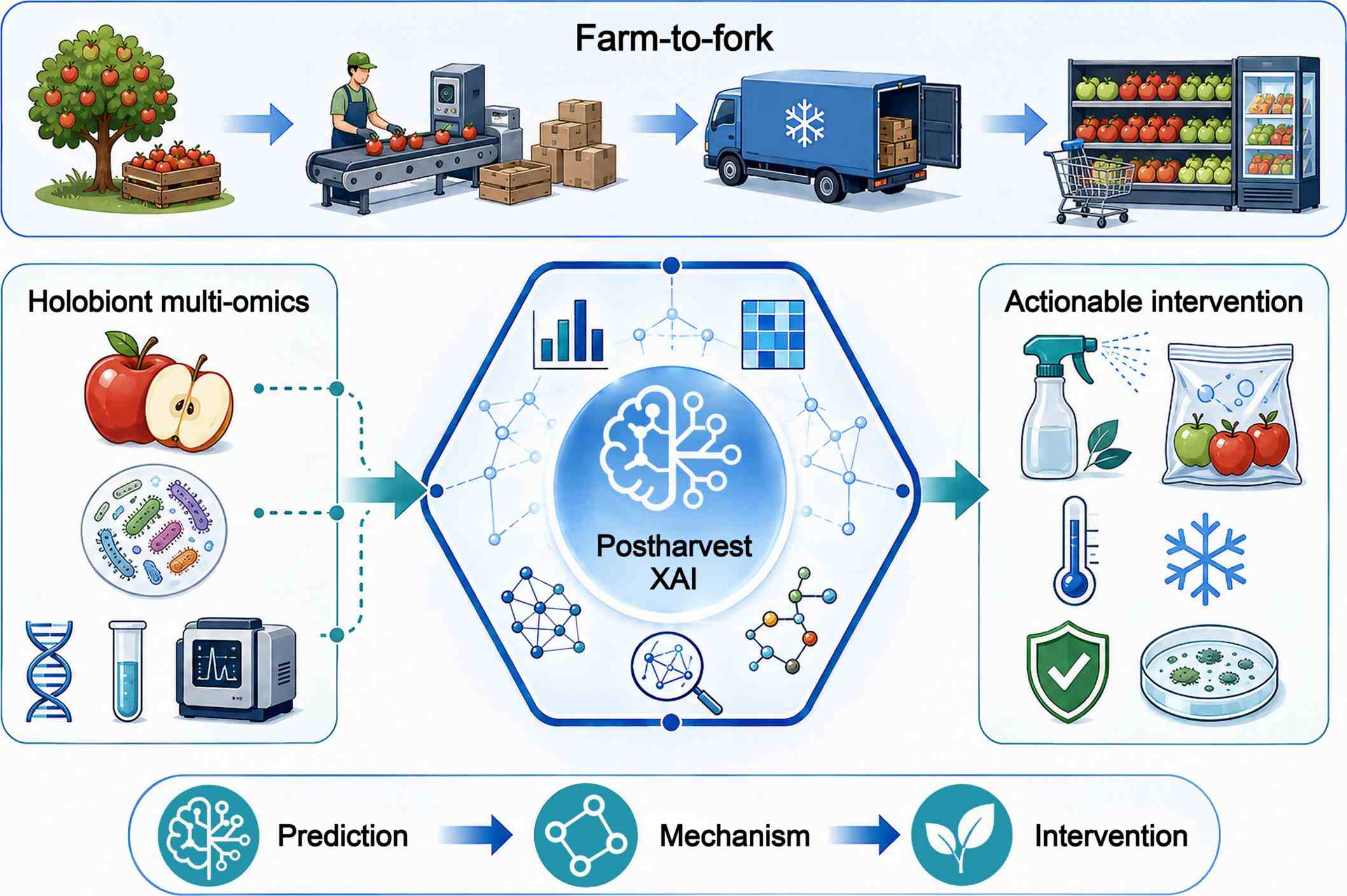
This study presents a cross-tissue, single-cell/single-nucleus transcriptomic atlas spanning from prenatal to postnatal developmental stages in pig, characterizing 83 distinct cell types from 252,033 cells. Through the construction of cross-tissue transcriptional regulatory networks and pseudotime trajectories, we revealed that organogenesis is driven by the coordinated processes of stem/progenitor cell proliferation, lineage specification, and functional maturation. Trajectory analysis further uncovered a conserved transcriptional bifurcation pattern associated with immune cell diversification. By integrating population genomics data, a muscle-specific enhancer near the MYOT gene was identified that shows signatures of selection and is associated with divergence in meat quality. Cross-species comparisons confirmed substantial cellular conservation with humans. Collectively, this atlas serves as a key resource for understanding mammalian development, informing precision breeding strategies, and advancing the pig as a biomedical model.
Microbial keystone taxa and metabolic signatures in centenarians regulate intestinal homeostasis during aging
- 19 May 2026
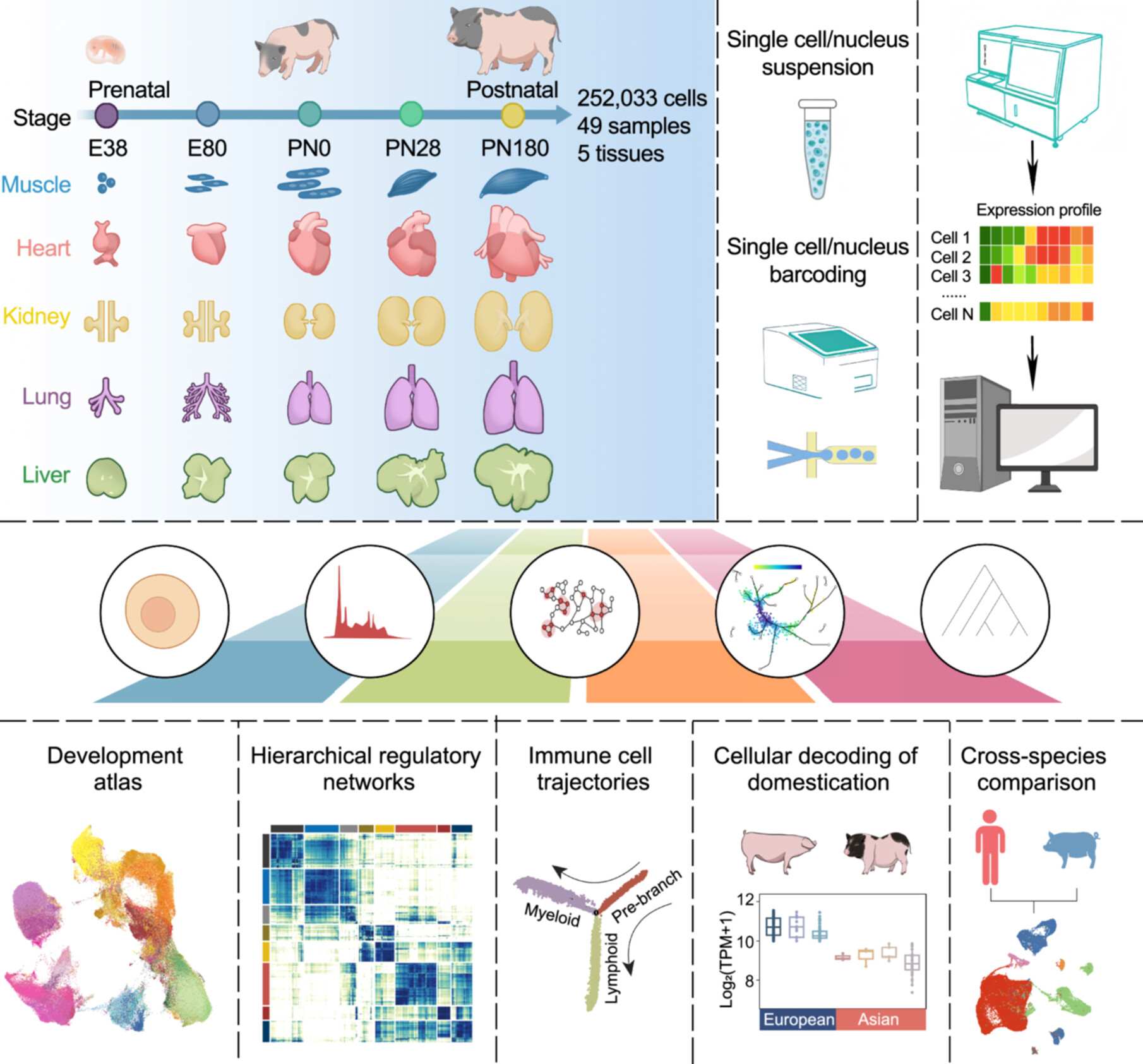
Clostridium scindens and its metabolite indole-3-acetic acid (IAA) are identified as a keystone taxon and metabolic signature in centenarians. Clostridium scindens enhances microbial network stability and produces IAA via its own amidase (AMIE) and aldehyde dehydrogenase (ALDH), thus promoting intestinal homeostasis by activating the aryl hydrocarbon receptor (AHR)-mediated Claudin-10 signaling in aged mice.
Spinal cord injury induces acute microbiome shock and system-wide transcriptomic reprogramming
- 03 May 2026
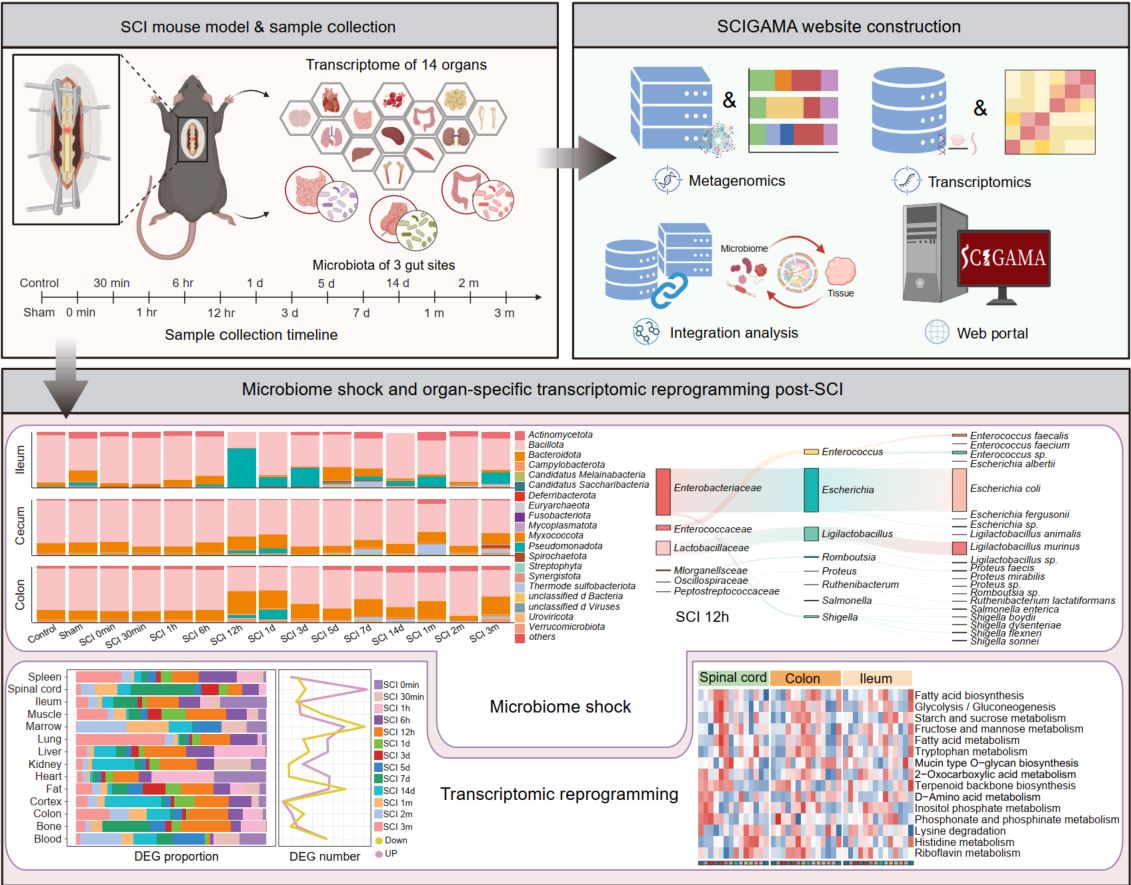
This study investigates the systemic consequences of spinal cord injury (SCI), with a particular focus on alterations in the gut microbiome and multi-organ transcriptomic responses. We identify a rapid and severe disruption of the gut microbiota—termed “microbiome shock”—that emerges within 12 h post-SCI and persists before gradually resolving by 5 days post-injury. To support further research in this field, we established an open-access resource, the Spinal Cord Injury Gut Microbiome and Multi-Organ Gene Expression Atlas (SCIGAMA).
Patient-derived organoid-immune co-cultures integrated with multi-omics reveal immunotherapy resistance mechanisms in urothelial carcinoma
- 07 May 2026
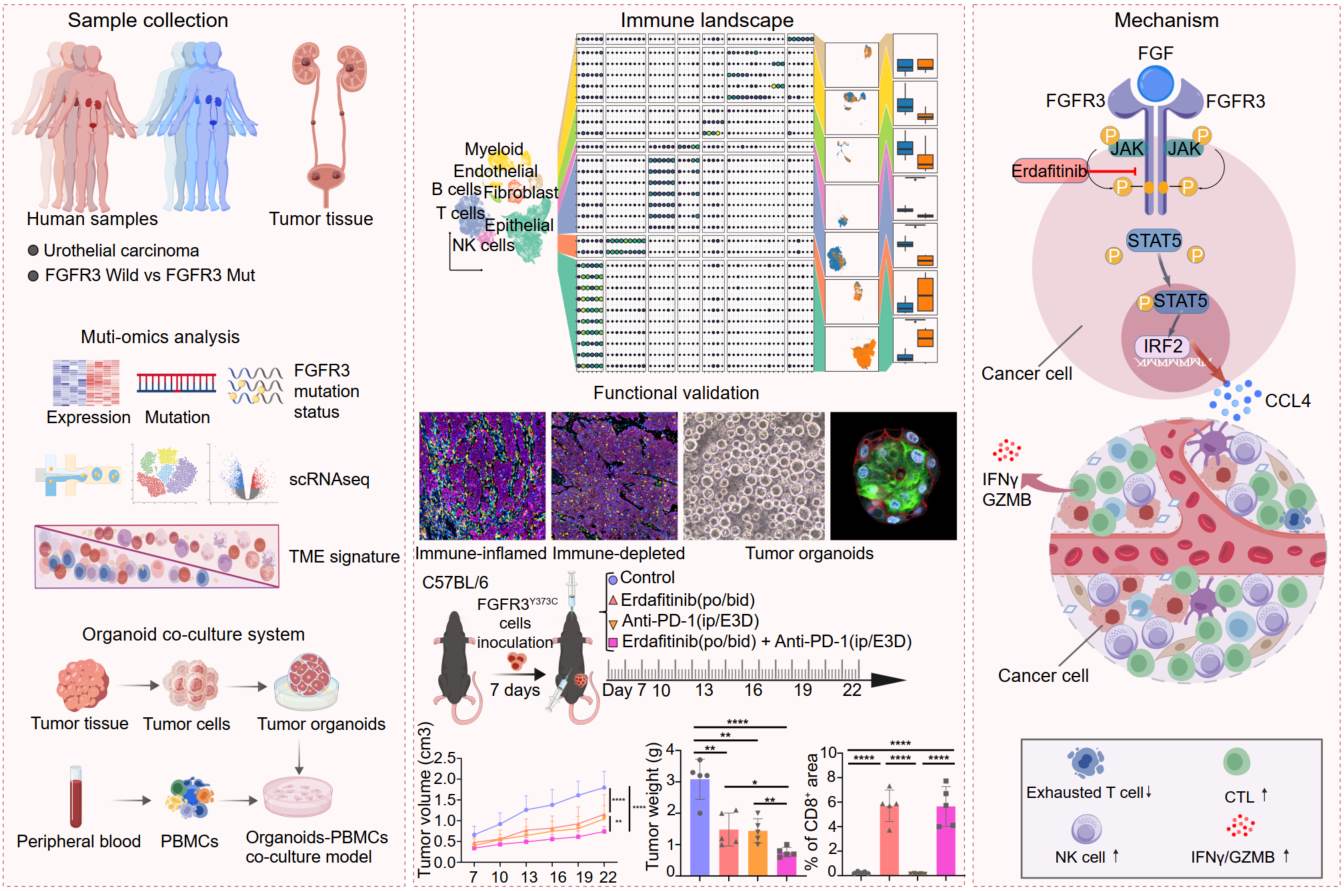
Immunotherapy resistance presents a formidable challenge in tumor biology. While fibroblast growth factor receptor 3 (FGFR3) serves as a pivotal oncogenic driver in a multitude of cancers, the exploration of its role in immune checkpoint inhibitor (ICI) resistance remains scarce, thus impeding a deeper understanding of the tumor immune microenvironment (TIME) in the era of immunotherapy. Employing patient-derived urothelial carcinoma (UC) organoids and co-cultured systems, along with single-cell RNA sequencing (scRNA-seq), whole-exome sequencing (WES), bulk RNA-seq, and CUT& Tag epigenomics in UC cohorts, we identified and characterized the key downstream mediators of FGFR3. The TIME associated with FGFR3 mutations exhibited a depletion of NK and CD8+ T cells, while simultaneously harboring an accumulation of exhausted effectors, correlating with diminished ICI response. Erdafitinib reprogrammed this “cold” TME into an inflamed state through a novel FGFR3–STAT5–IRF2 signaling cascade. These findings, corroborated by a wealth of evidence, advocate for the combination of FGFR3-targeted therapy with immunotherapy for UC, bridging critical pre-clinical and clinical insights.
Integrative host–microbiome modeling uncovers the implication of oral–gut translocation in advanced cirrhosis
- 29 April 2026
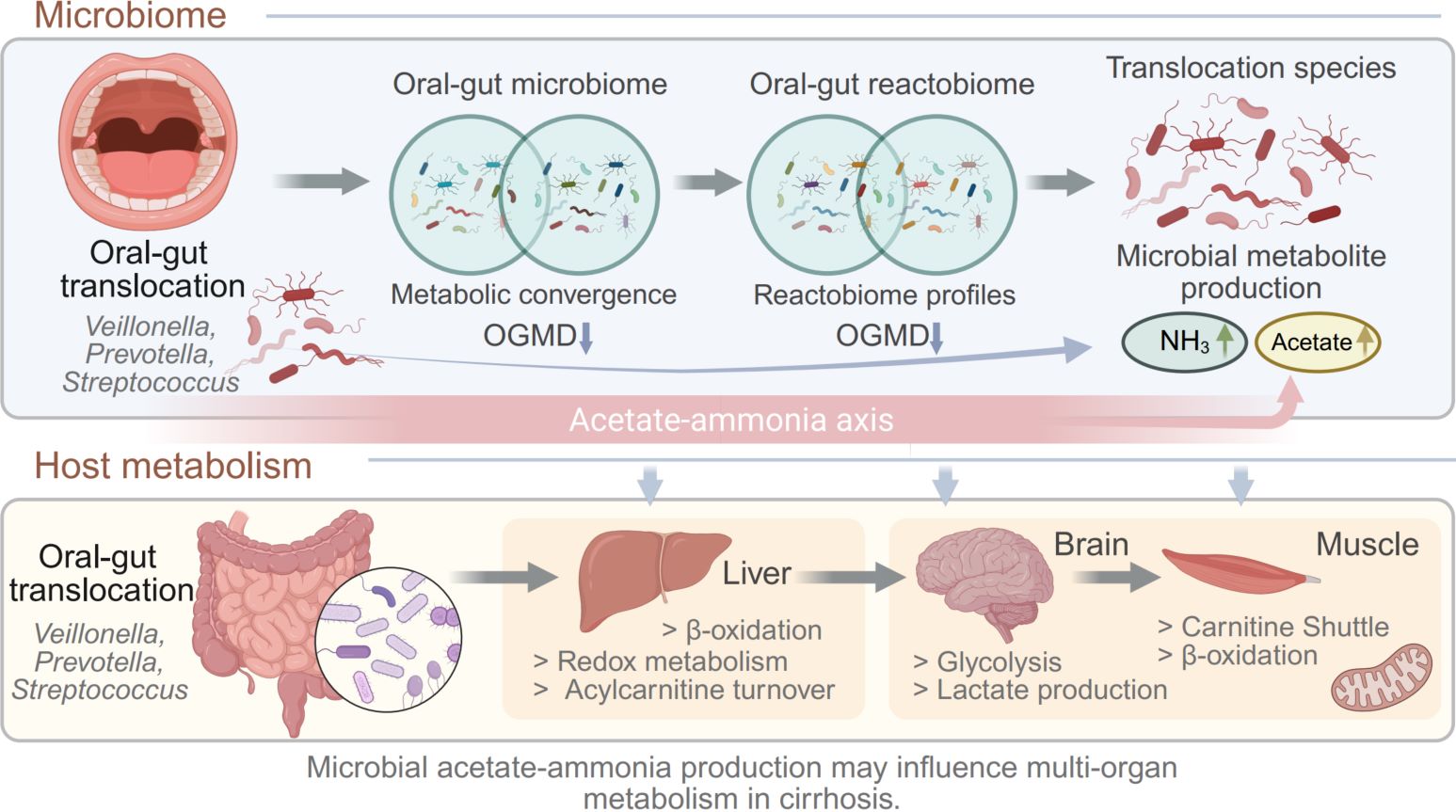
Liver cirrhosis is associated with profound disruption of host–microbiome metabolic interactions. Using paired oral and fecal metagenomics combined with genome-scale metabolic modeling, we investigated how microbial translocation along the oral–gut axis influences microbial metabolism at different cirrhosis severities. Reactobiome-based functional profiling revealed progressive metabolic convergence between oral and gut microbiomes, quantified by a decrease in oral–gut metabolic distance. Translocation-associated microbial species enriched in patients with cirrhosis were predicted to have elevated capacities for ammonia and acetate production. Microbial-community and host metabolic modeling further suggested that these microbial metabolic shifts may influence host energy metabolism and redox balance across the liver, brain, and skeletal muscle. Together, these findings suggest a potential acetate-ammonia metabolic axis linking oral–gut microbial translocation with systemic metabolic stress in advanced cirrhosis.
Microbial synthetic community ARC prevents aflatoxin and increases rhizobia-legume nodulation couplingly
- 29 April 2026
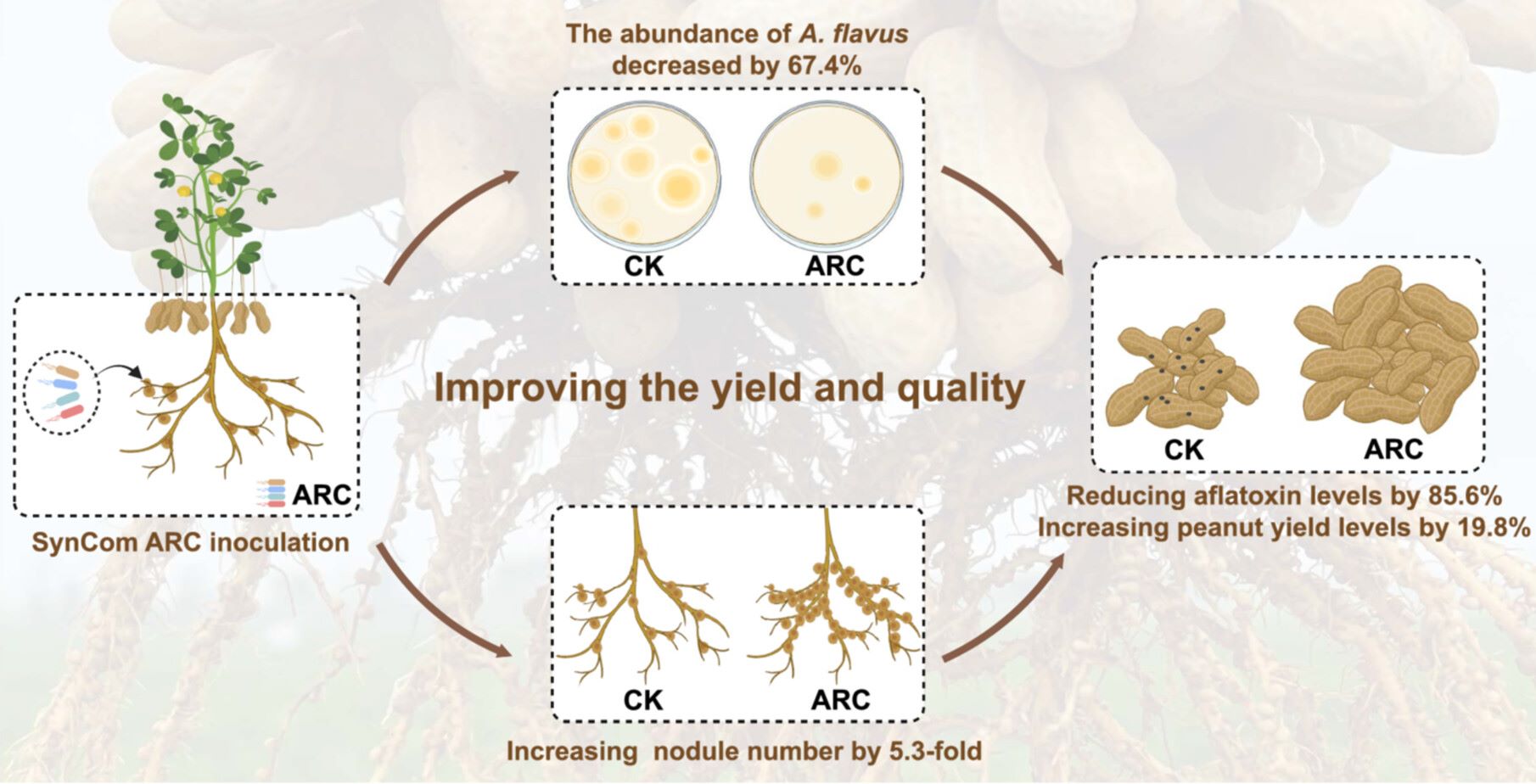
Develop a novel strategy for exploring a dual-functional microbial synthetic community. Invent the SynCom ARC, which achieves aflatoxin control and rhizobia nodulation induction coupling in peanut. SynCom ARC inhibits A. flavus growth and reduces peanut aflatoxin levels by 85.6% in 4 year field trials. SynCom ARC enhances peanut nodulation and nitrogenase activity, retains active nodules at harvest, and boosts yield without super nodulation penalty in 325 sites of 19 provinces. SynCom ARC inhibits multiple targets in A. flavus, recruits and activates nodulation and nitrogen fixation in rhizobia and peanut, and improves photosynthesis and carbon supply for aflatoxin prevention and nodulation induction, balancing yield increase.
Single-cell transcriptomics reveals cellular heterogeneity and phenotypic transitions of smooth muscle cells in aortic dissection
- 21 April 2026
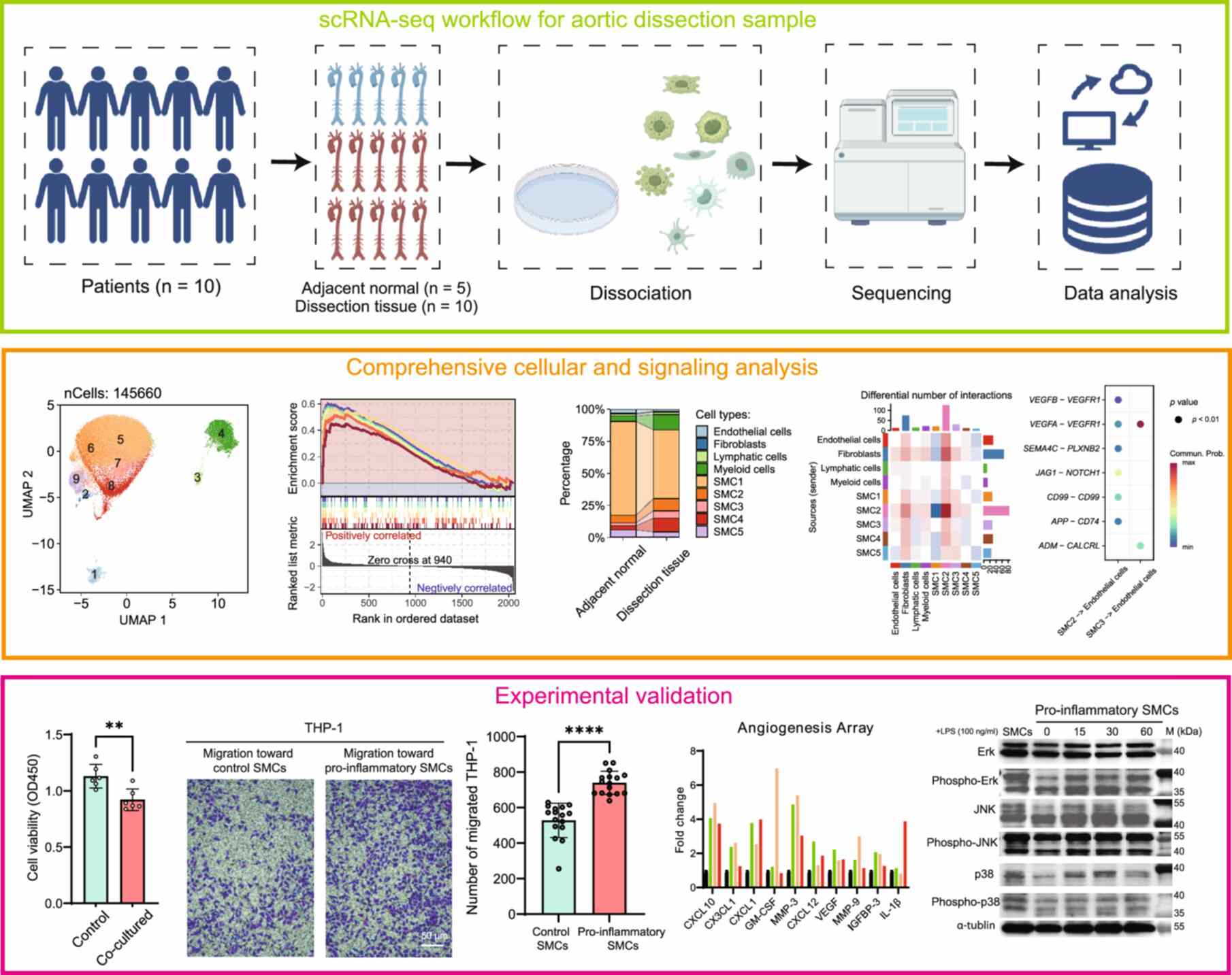
We utilized single-cell RNA sequencing (scRNA-seq) to investigate cellular heterogeneity and signaling networks in aortic dissection (AD) tissues compared to adjacent normal tissues. The analysis identified five smooth muscle cell (SMC) subtypes, with SMC2 linked to fibrosis and SMC3 associated with inflammation. Thrombus-positive AD samples showed upregulated angiopoietin-like 4 (ANGPTL4) and increased M2 macrophages, indicating an immunosuppressive microenvironment. Cell-cell communication analysis revealed a shift in vascular endothelial growth factor A (VEGFA) signaling from SMCs to fibroblasts, disrupting vascular homeostasis. In vitro experiments confirmed SMC2-induced endothelial-to-mesenchymal transition and SMC3-driven inflammatory responses via mitogen-activated protein kinase (MAPK) pathways. Immunofluorescence validated elevated insulin-like growth factor binding protein 2 (IGFBP2), procollagen-lysine 2-oxoglutarate 5-dioxygenase 2 (PLOD2), and VEGFA in AD tissues, supporting their roles in matrix remodeling and angiogenesis. These findings highlight SMC phenotypic switching and altered VEGFA signaling as key drivers of AD, proposing novel therapeutic targets to restore vascular integrity.
FluNexus: A versatile web platform for antigenic prediction and visualization of influenza A viruses
- 02 May 2026
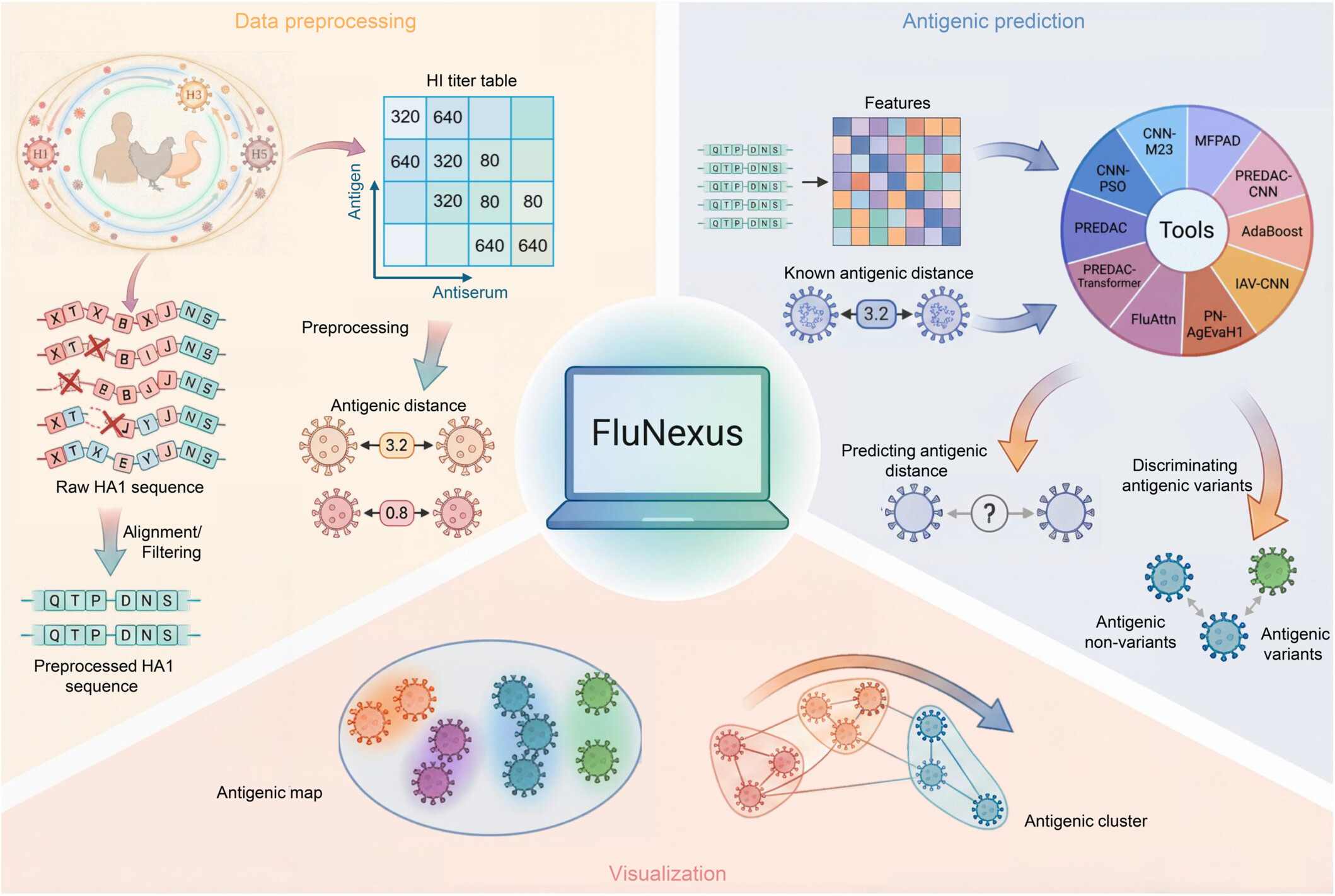
FluNexus is a versatile platform for the antigenic prediction and visualization of influenza A viruses, including: (i) Online data preprocessing module. (ii) Online antigenic prediction module. (iii) Visualization module for mapping antigenic evolution.
Intratumoral Enterobacter hormaechei drives gemcitabine resistance in pancreatic cancer via cddL-mediated drug inactivation
- 29 April 2026
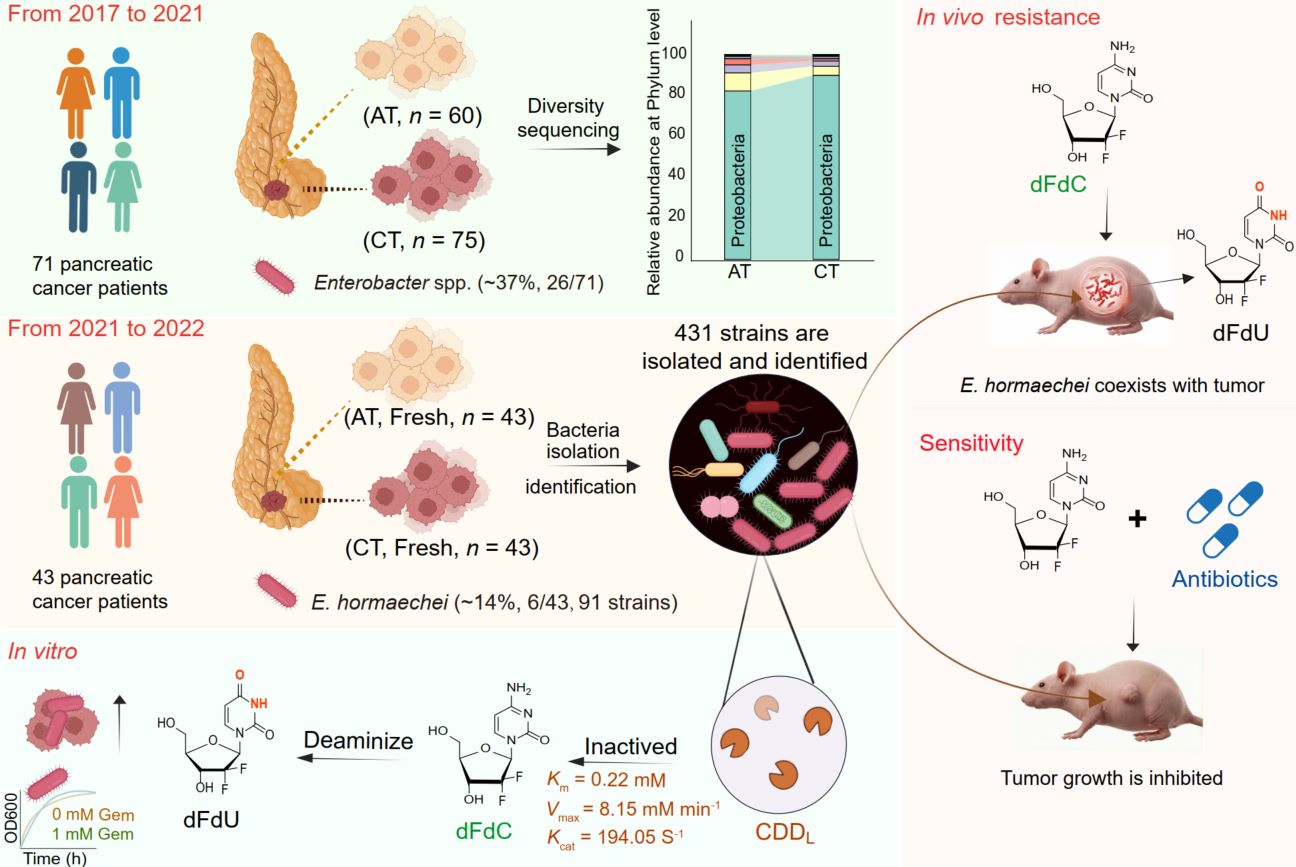
Gemcitabine resistance poses a critical barrier to improving survival in pancreatic cancer, yet the microbial drivers remain elusive. By integrating 16S rRNA amplicon sequencing with large-scale culturomics across 114 clinical samples, we identified Enterobacter hormaechei as a key intratumoral pathogen. We demonstrate that E. hormaechei confers resistance by enzymatically converting the drug to its inactive metabolite dFdU via a unique long-isoform cytidine deaminase encoded by cddL. Kinetic analysis revealed exceptional catalytic efficiency (Km = 0.22 mM, kcat = 194.05 s−1), and genetic ablation of cddL fully restored drug sensitivity. In vivo, antibiotic co-treatment eliminated intratumoral bacteria and potentiated gemcitabine efficacy, enabling a 50% dosage reduction without comprising therapeutic outcome. Pan-cancer analysis further confirmed the broad prevalence of Enterobacter across multiple solid tumor types. These findings elucidate a cddL-mediated mechanism of chemoresistance and identify intratumoral E. hormaechei as a tractable therapeutic target for optimizing gemcitabine-based regimens and improving patient outcomes.
Navigating the future of nano-pesticides: A perspective on design, efficacy, mechanisms, and environmental stewardship
- 29 April 2026
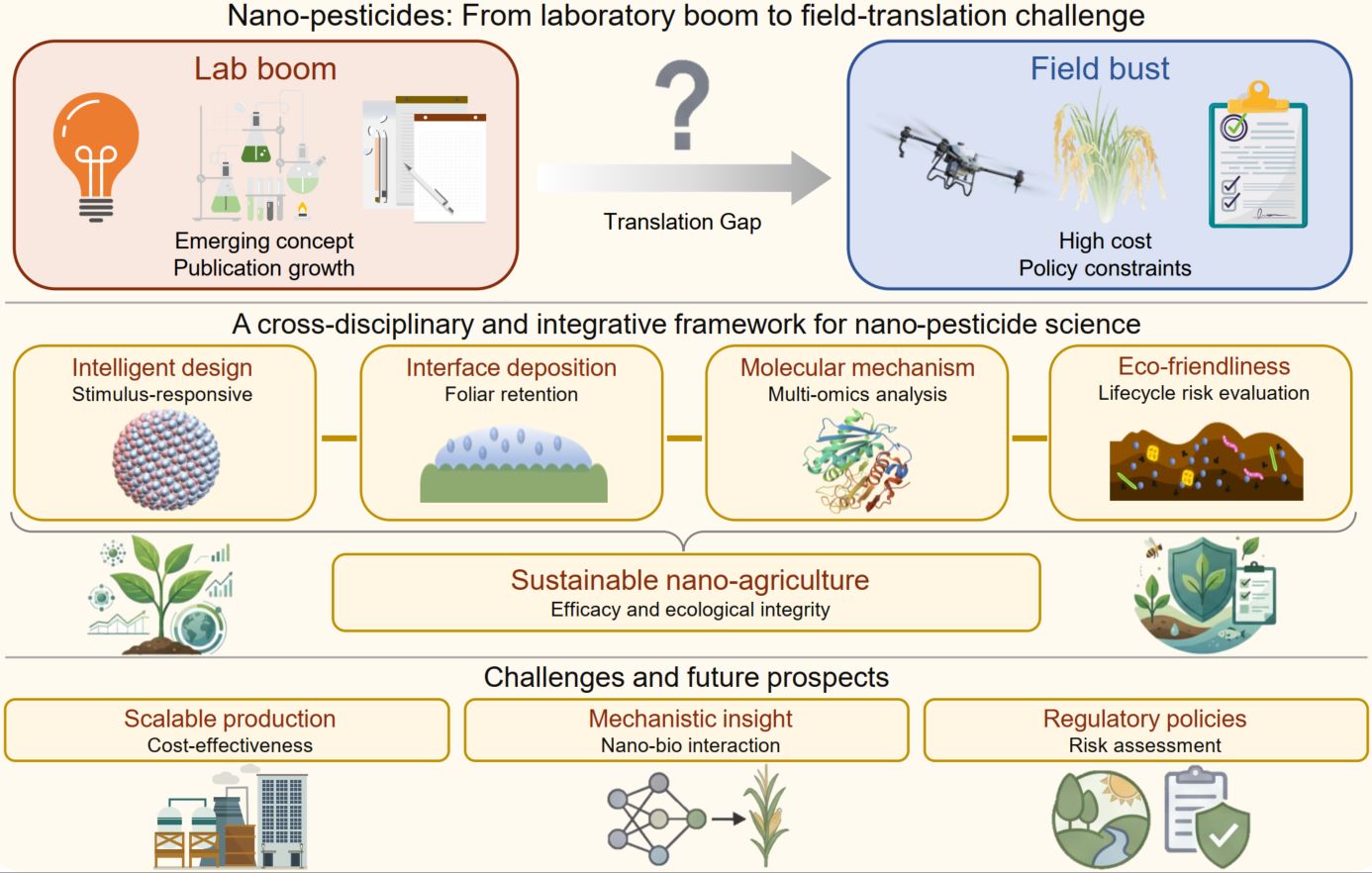
Nano-pesticides are driving a paradigm shift toward sustainable plant protection. This review systematically synthesizes recent advances across four interconnected domains: (i) intelligent formulation design for targeted delivery and controlled release; (ii) interfacial behavior regulation to enhance foliar deposition; (iii) multi-omics elucidation of synergistic efficacy and molecular interactions; and (iv) environmental fate management, including risk mitigation and microbiome remediation. By integrating these multiscale innovations, the field is advancing nano-enabled crop protection from laboratory research toward field application. Collectively, this convergence provides a scientific foundation and interdisciplinary roadmap needed to build next-generation agricultural systems that are resource-efficient and ecologically compatible.
Chromatin unwinding state for in situ identification and lineage tracing of circulating tumor cells
- 09 March 2026
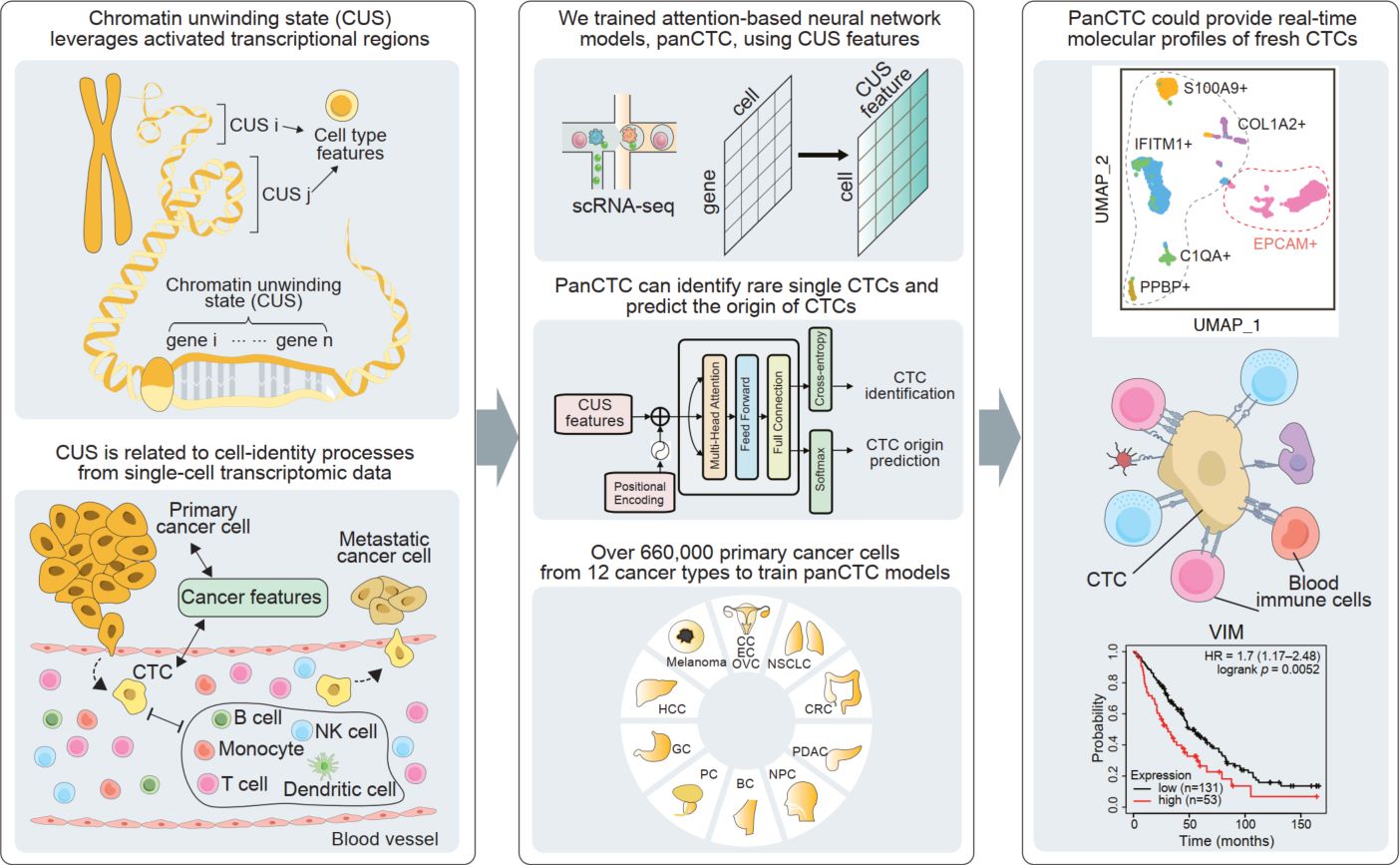
The detection of circulating tumor cells (CTCs) through liquid biopsy offers a non-invasive approach for accurately monitoring cancer dissemination and evaluating therapeutic efficiency. However, their rarity and heterogeneity limit conventional tumor antigen labelling-based methods in identifying and tracing CTCs. Here, we developed a novel metric, termed chromatin unwinding state (CUS), which leverages activated transcriptional regions related to cell-identity processes from single-cell transcriptomic data while overcoming technical variances. Using CUS features, we trained attention-based neural network models, panCTC, to in situ identify and lineage trace rare single CTCs directly from 5 mL of peripheral blood mononuclear cells scRNA-seq without enrichment. We benchmarked panCTC on various in silico-simulated, public, and in-house sequenced data, demonstrating its robustness across sample types and platforms. PanCTC could provide real-time scRNA-seq profiles of fresh CTCs, supporting early cancer detection and targeted anti-metastatic therapy.
Spatial multi-omics identifies a NOTCH3-mediated capillary–mCAF crosstalk driving immune exclusion in hepatocellular carcinoma
- 17 March 2026
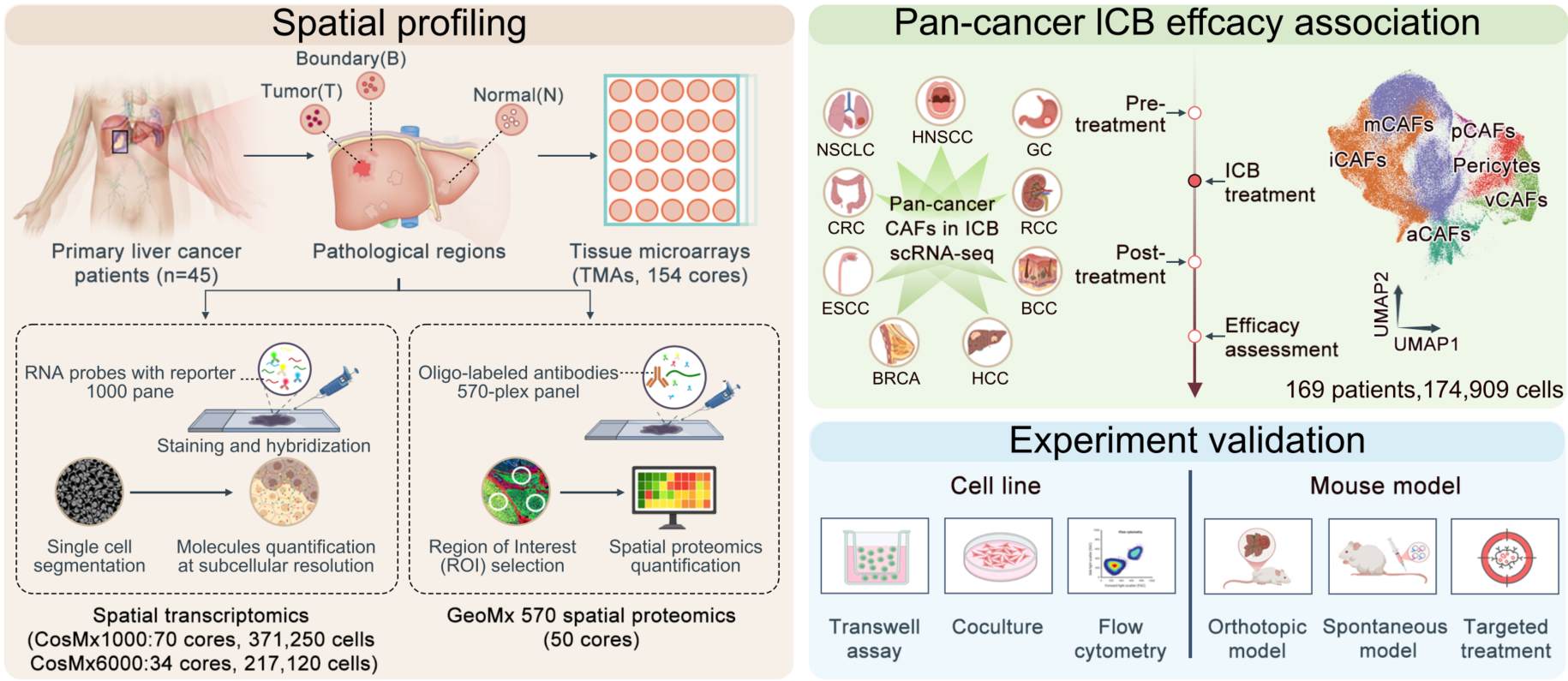
Spatial multi-omics defines a DLL4–NOTCH3–driven capillary–mCAF axis orchestrating fibrotic remodeling and immune exclusion in hepatocellular carcinoma tumor cores, conferring immune checkpoint blockade resistance. Targeting stromal NOTCH3 disrupts this pro-fibrotic niche, enhances T cell infiltration, and synergizes with anti-PD-1 therapy, highlighting a translatable strategy to overcome immunotherapy resistance.
Two stable gut microbiome guilds predict liver tumor class and treatment responses
- 02 April 2026
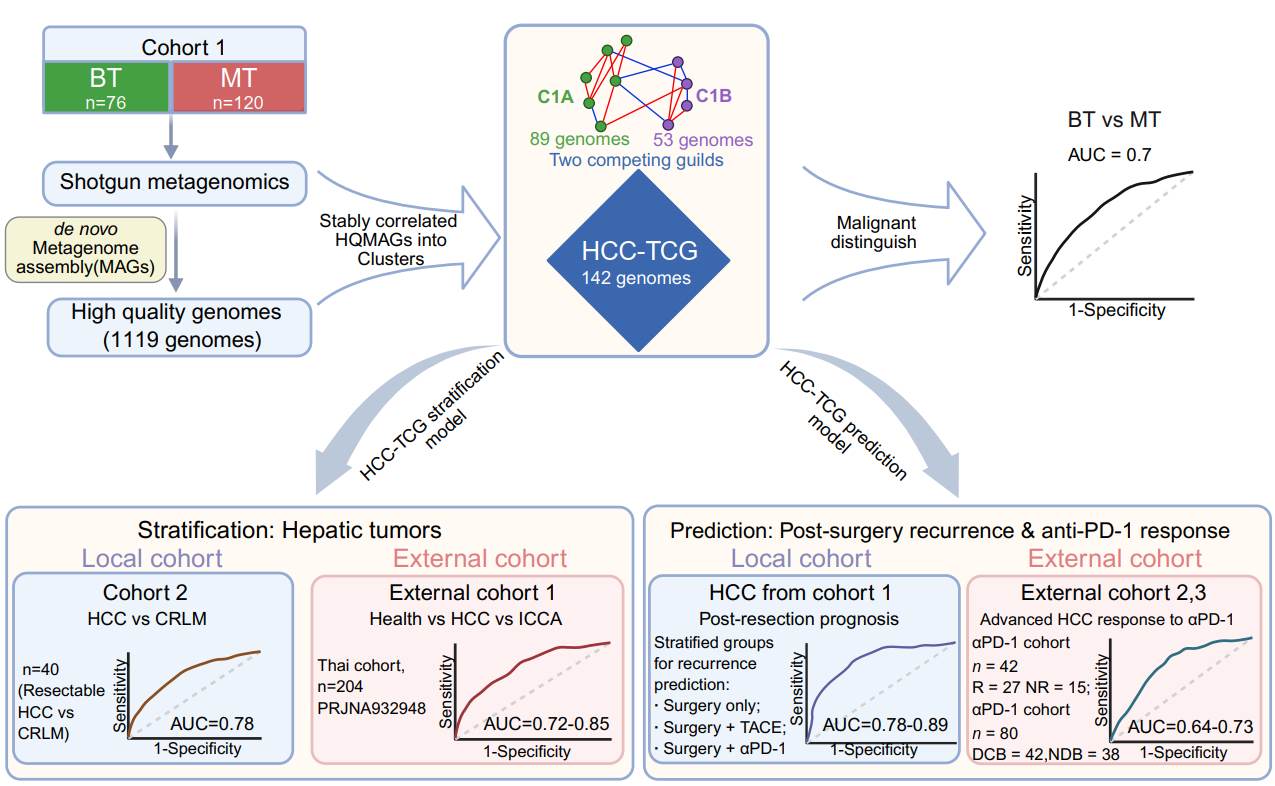
Gut microbiome alterations are linked to hepatocellular carcinoma (HCC), yet taxon-based biomarkers often lack reproducibility. Using shotgun metagenomics from 120 resectable HCC (MT) and 76 benign hepatic tumor controls (BT), we reconstructed a stable interaction-based Two Competing Guilds model (HCC-TCG) comprising 142 genomes organized into two antagonistic guilds. The ecological guild balance distinguished HCC from benign and other hepatic tumor types, generalized across external cohorts, and predicted post-resection recurrence and adjuvant therapy response. Validation in advanced HCC immunotherapy cohorts further supported its transferability, establishing a genome-resolved ecological signature for tumor stratification and noninvasive risk prediction.
A large-scale single-cell transcriptomic atlas indicates the immune panorama of influenza A infection
- 25 March 2026
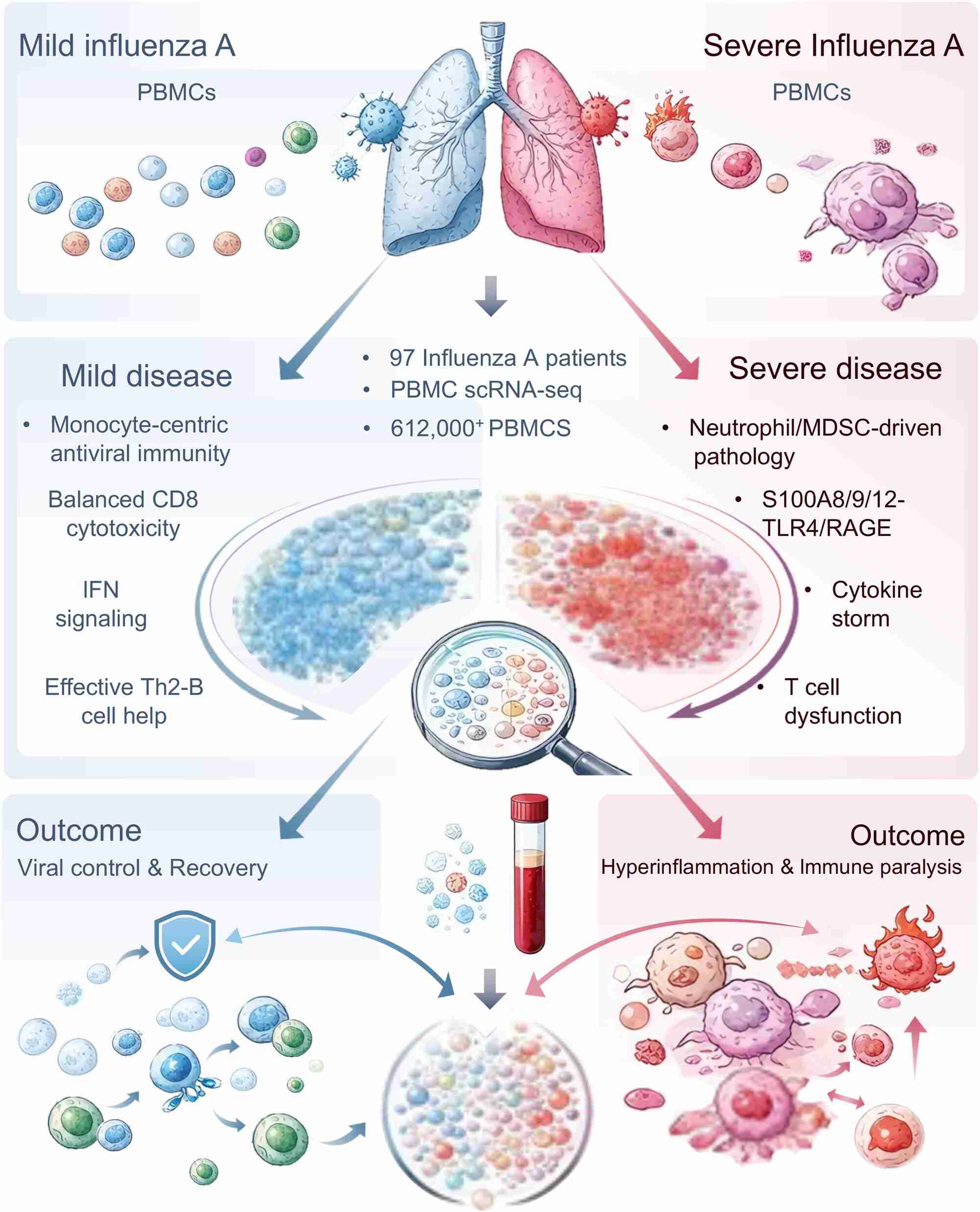
The study presents a large-scale single-cell transcriptomic atlas profiling over 612,010 peripheral immune cells from 97 individuals to decode the heterogeneity of influenza infection. These findings indicate a fundamental immune dichotomy determining clinical trajectories: a protective, monocyte-centric antiviral state in mild disease versus a pathological, neutrophil- and myeloid-derived suppressor cell (MDSC)-driven hyperinflammatory state in severe infection. Mechanistically, severe disease is orchestrated by the S100A8/9/12–TLR4/RAGE axis and MDSC expansion, driving a “double hit” of cytokine storm and immune paralysis. This dysregulated environment precipitates metabolic collapse in effector T cells and the pathogenic conversion of regulatory T cells. Collectively, these results define the cellular checkpoints governing influenza outcomes and highlight host-directed therapeutic targets.
Targeting DGKα/PA axis inhibits tumor immune evasion and augments sensitivity to immunotherapy in gastrointestinal cancers
- 12 March 2026
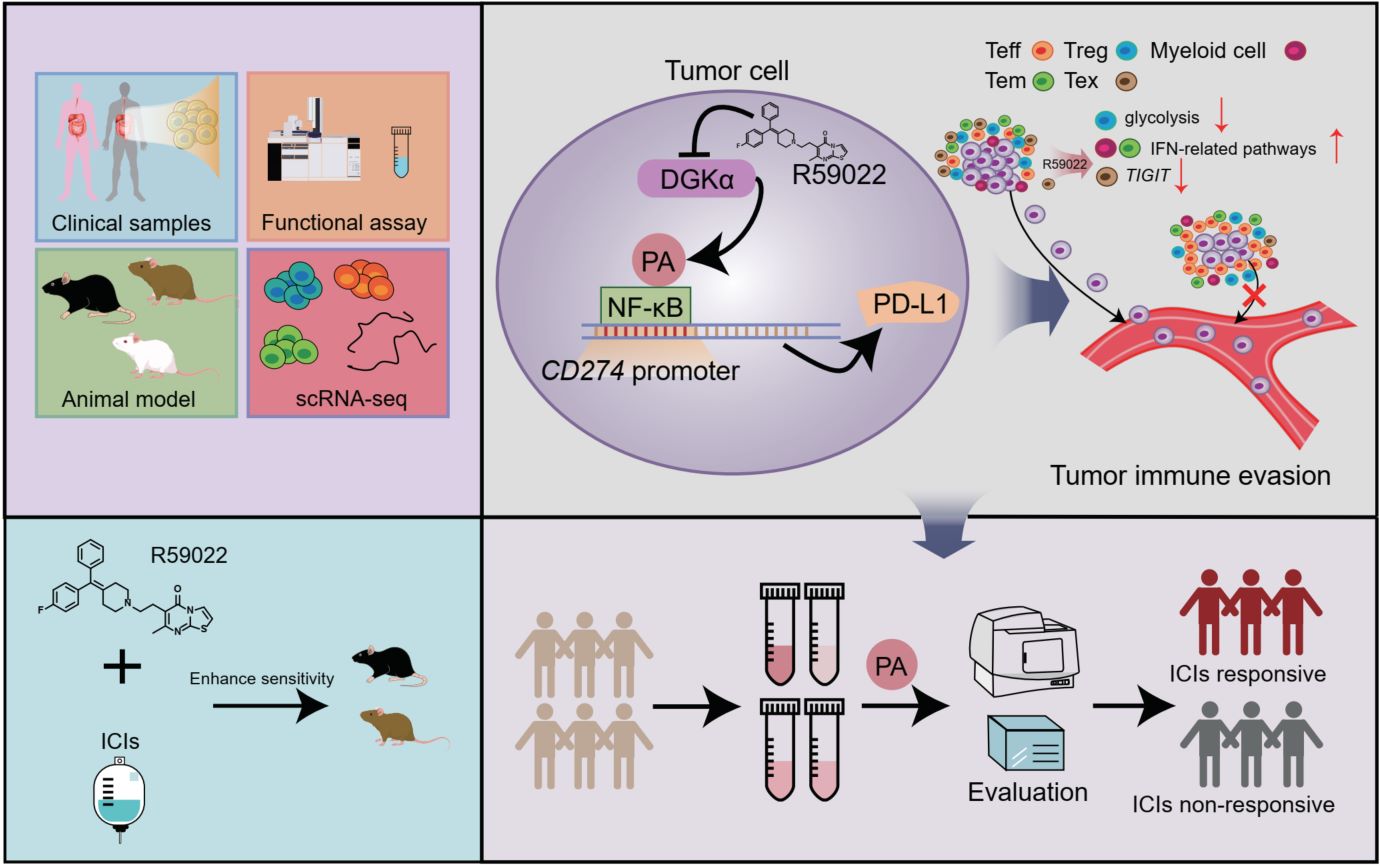
Immune checkpoint inhibitors (ICIs) have shown promising antitumor efficacy in certain types of solid tumors. However, the efficacy of ICIs remains unsatisfactory owing to the dysregulation of signaling pathways in local tumor tissues. Here, we reveal that diacylglycerol kinase α (DGKα)-derived phosphatidic acid (PA) directly binds to nuclear factor-κB (NF-κB) and enhances the transcriptional activity of NF-κB to increase the expression of programmed cell death 1-ligand 1 (PD-L1) and facilitate the immune evasion of tumor cells and orchestrate immune microenvironment. Inhibition of DGKα activity decreases the intratumoral PD-L1 level and induces cytotoxic T lymphocytes (CTLs) infiltration and resultantly enhances the antitumor efficacy of ICIs. Plasma PA can function as a biomarker to evaluate the efficacy of ICIs in gastrointestinal cancers. Overall, our results identify the DGKα/PA axis as a metabolic driver of immune evasion and CTLs exclusion, representing a promising target to enhance ICIs' efficacy in gastrointestinal cancer treatments.
Chondroitin sulfate restores muscle mass via gut–muscle axis remodeling through sugar–bile acid metabolism reprogramming
- 12 March 2026
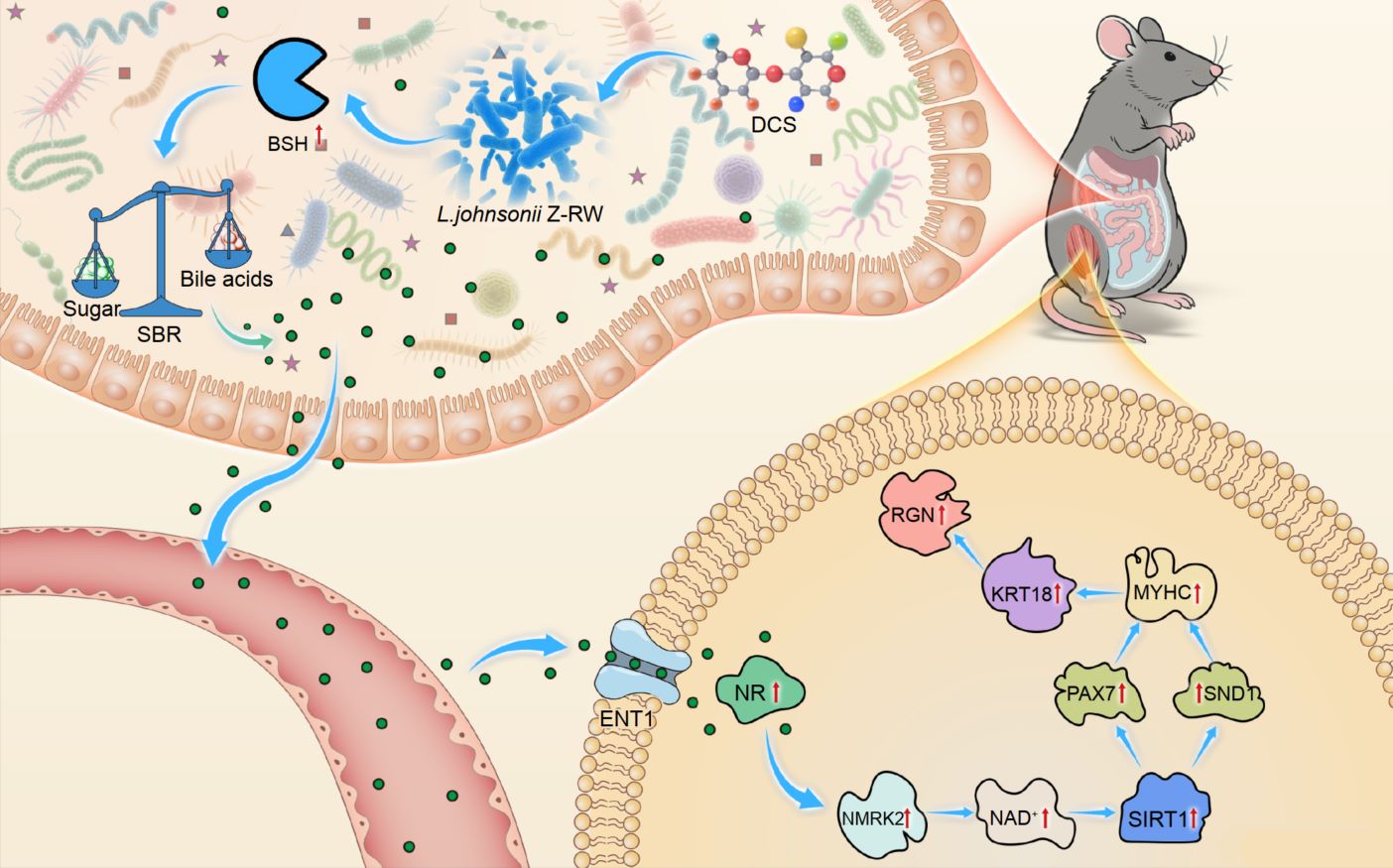
The graphical abstract illustrates the molecular mechanism by which chondroitin sulfate (DCS) alleviates glucocorticoid-induced myopathy through the gut–muscle axis. DCS selectively enriches L. johnsonii Z-RW, enhancing bshA-encoded BSH activity to promote bile acid deconjugation and reduce the sugar–bile acid ratio, thereby re-establishing intestinal metabolic homeostasis. The resulting secondary bile acids cross the intestinal barrier and reach muscle tissue, where they activate NMRK2-mediated NAD⁺ biosynthesis and PAX7 signaling, upregulating myogenic proteins such as KRT18, MYHC, and RGN. These coordinated molecular events restore energy metabolism and muscle function.
Accu16S/AccuITS: Accurate and broadly applicable amplicon sequencing for absolute microbiome quantification
- 02 March 2026
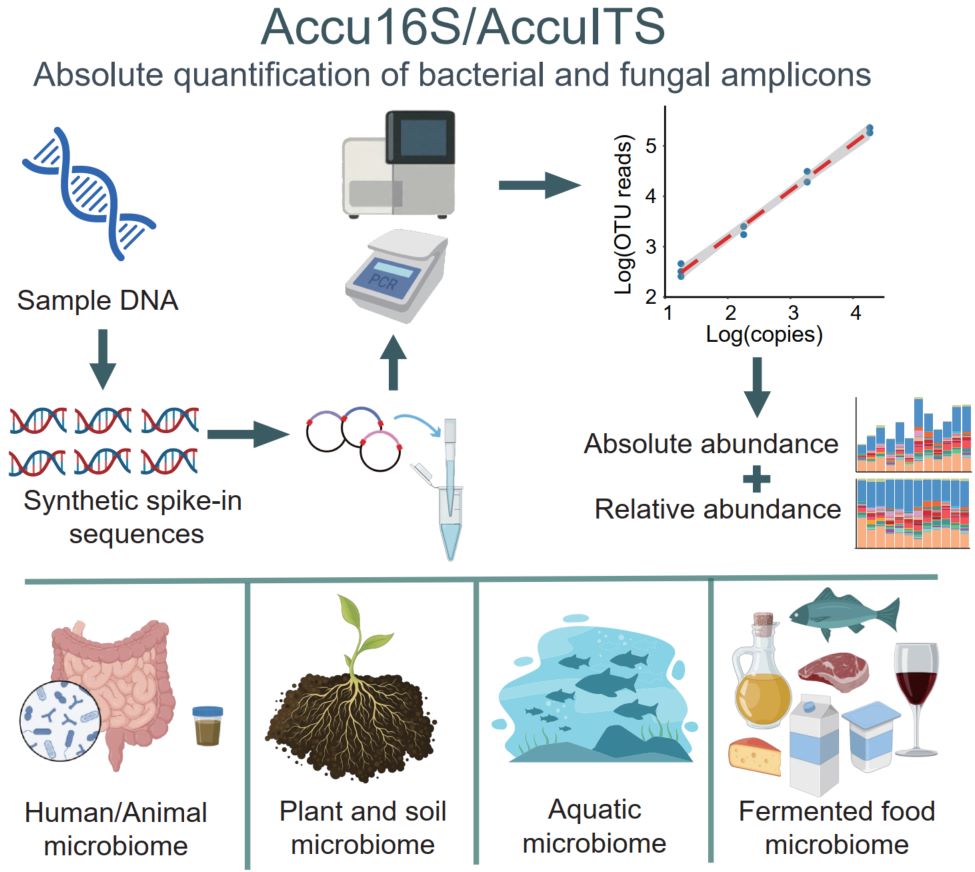
Traditional 16S rRNA gene and Internal Transcribed Spacer region amplicon sequencing provides only relative abundance, often leading to biased ecological interpretations. To overcome this limitation, we developed Accu16S/AccuITS, an absolute quantification method for bacterial and fungal amplicons based on synthetic internal spike-in DNA with known copy numbers. By adding internal standards prior to Polymerase Chain Reaction and sequencing, absolute microbial abundances can be calculated using standard curve regression. Accu16S/AccuITS exhibits sensitivity and consistency comparable to quantitative Polymerase Chain Reaction and is applicable to diverse sample types. A single sequencing run simultaneously yields relative abundance, total absolute abundance, and taxon-specific absolute abundance. Case studies across diverse ecosystems demonstrate that absolute quantification provides ecologically and functionally meaningful insights beyond those obtained from relative abundance analyses.
fastp 1.0: An ultra-fast all-round tool for FASTQ data quality control and preprocessing
- 09 September 2025
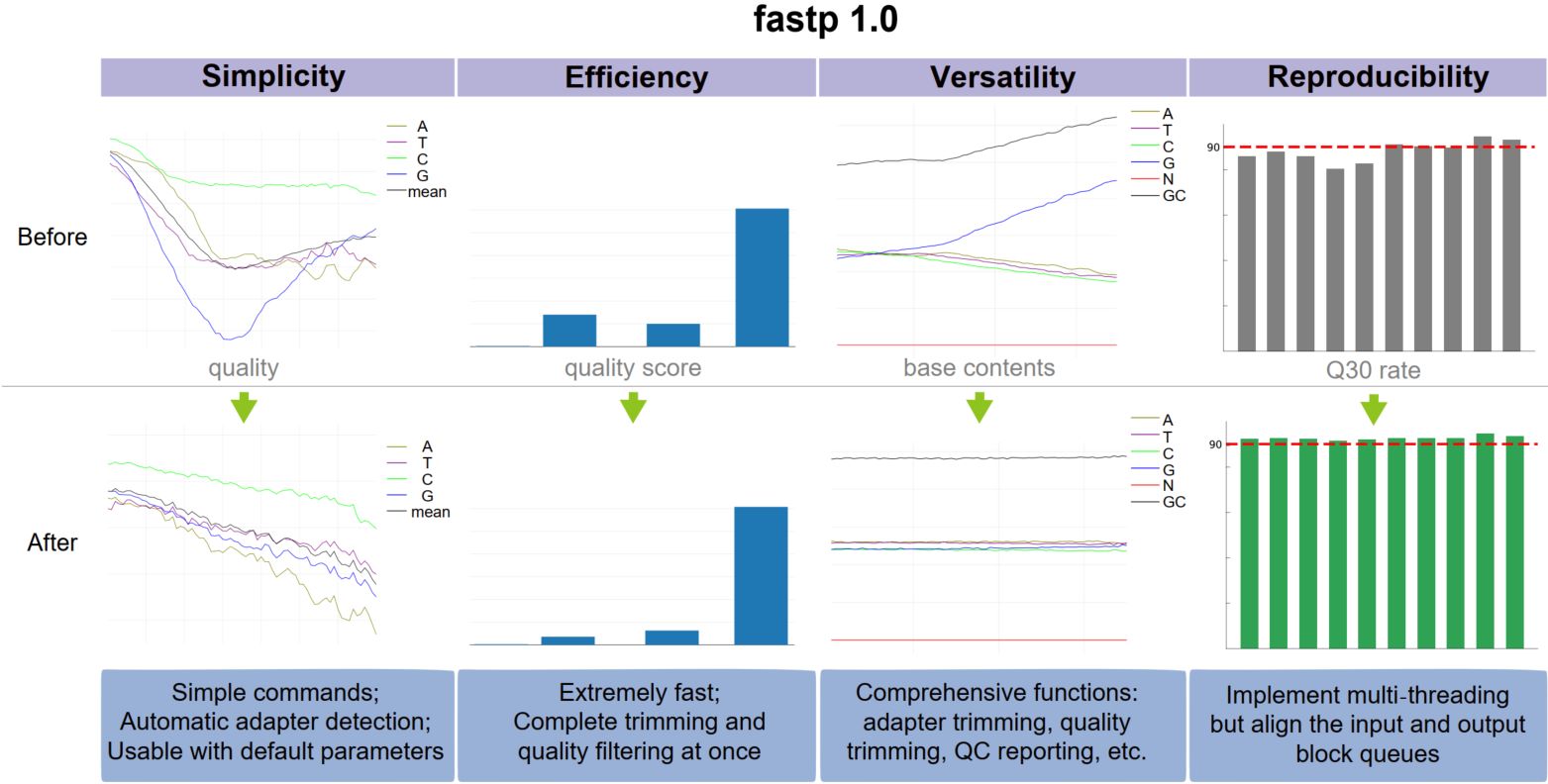
The figure shows side-by-side quality comparison of the data before and after preprocessing using fastp 1.0. As can be seen from this figure, the data quality has been significantly improved after fastp processing. The Q20 ratio and Q30 ratio increased, and the GC base ratio returned to balance.
EasyMetagenome: A user-friendly and flexible pipeline for shotgun metagenomic analysis in microbiome research
- 14 February 2025
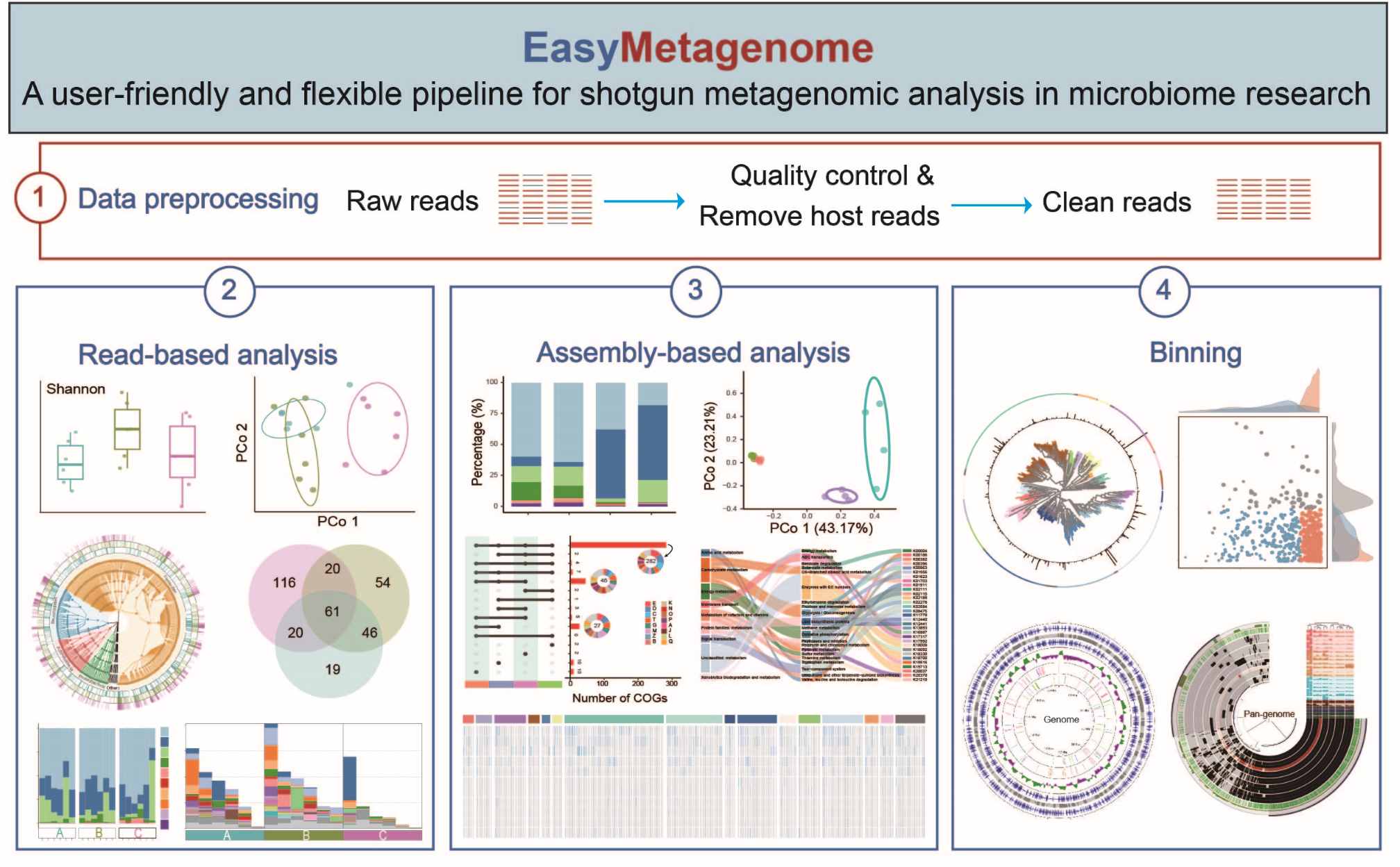
EasyMetagenome is a user-friendly shotgun metagenomics pipeline designed for comprehensive microbiome analysis, supporting quality control, host removal, read-based, assembly-based, binning, genome and pan-genome analysis. It offers customizable settings, data visualizations, and parameter explanations. The pipeline is freely available at https://github.com/YongxinLiu/EasyMetagenome.
Gut–X axis
- 26 February 2025
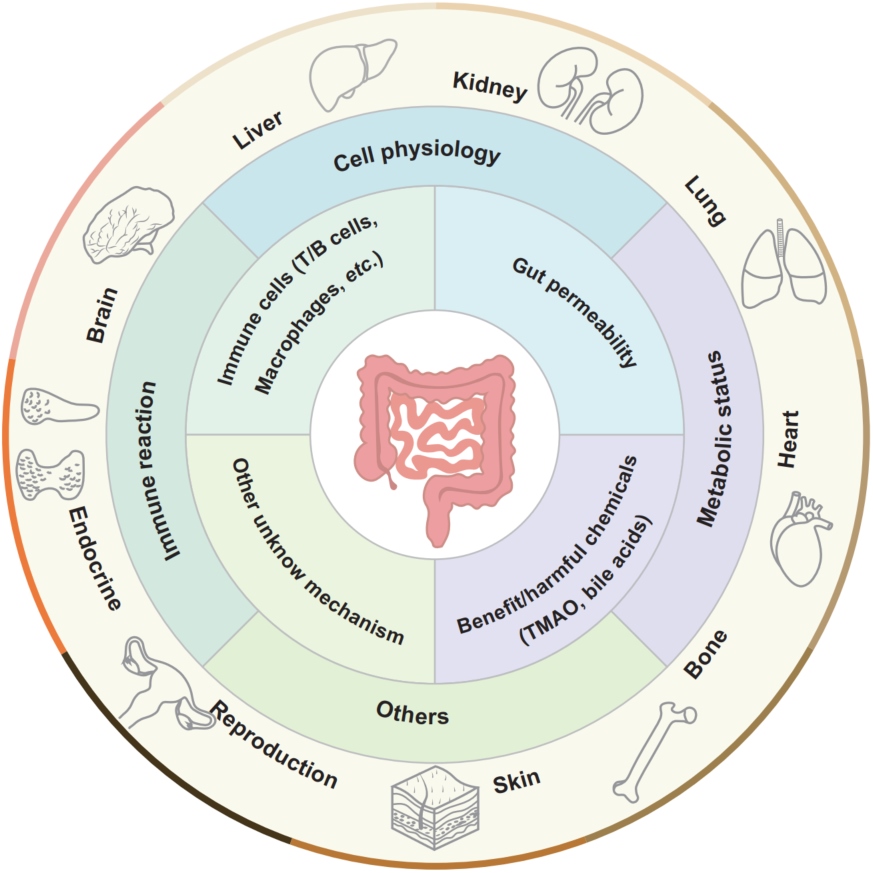
The concept of “gut–X axis”: the intestine and intestinal microbiota are proven to be able to modulate the pathophysiologic progressions of the extraintestinal organs' diseases. The bioactive chemicals and/or intestinal immune cells can translocate into the circulatory system and other organs and influence the immune reactions, metabolic status, cells physiology, and so forth of extraintestinal organs, finally regulating these organs' homeostasis. Meanwhile, other organs may reversely impact the intestine, namely such regulatory axis is bidirectional.
Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp
- 08 May 2023
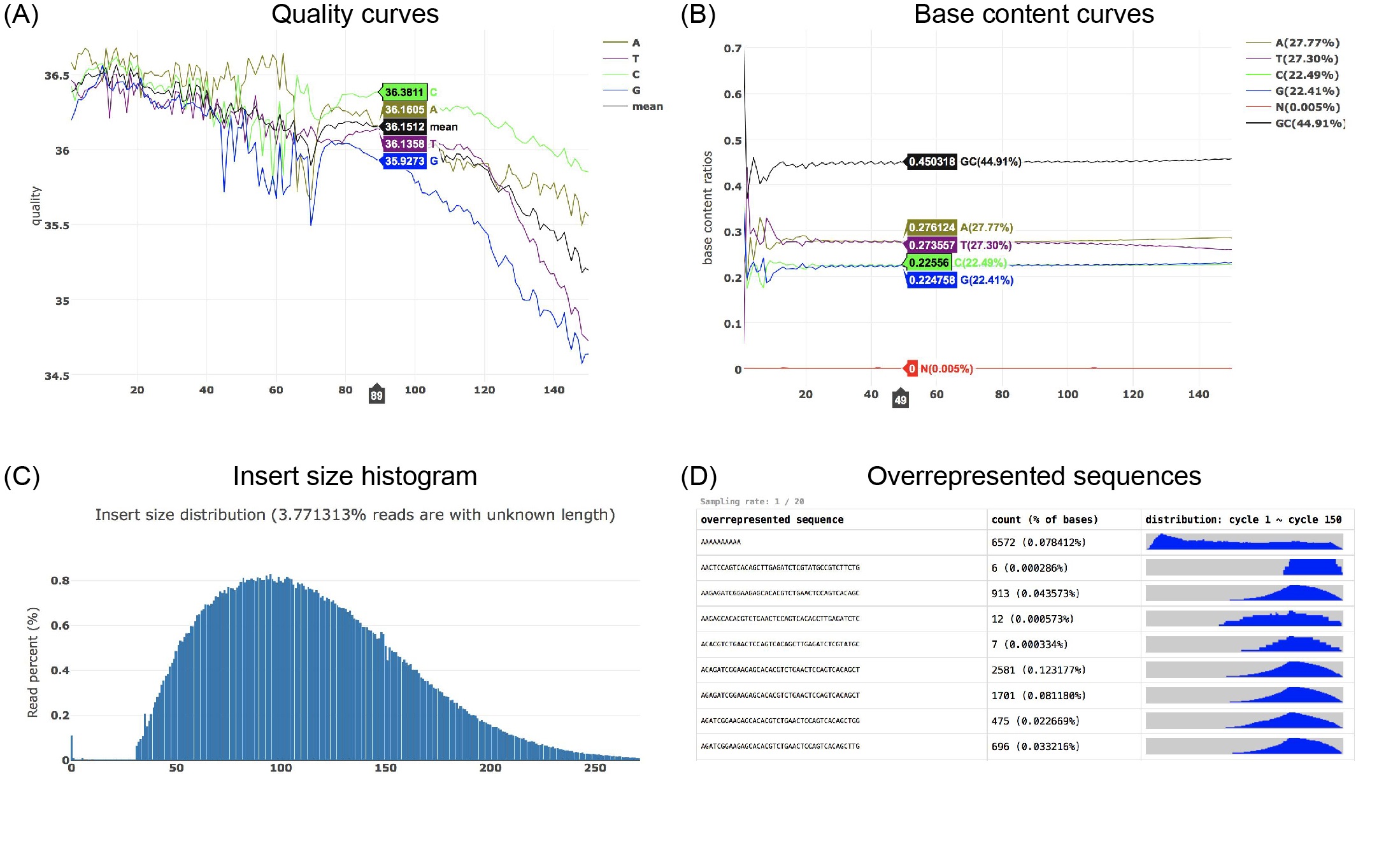
Fastp is a widely adopted tool for FASTQ data preprocessing and quality control. It is ultrafast and versatile and can perform adapter removal, global or quality trimming, read filtering, unique molecular identifier processing, base correction, and many other actions within a single pass of data scanning. Fastp has been reconstructed and upgraded with some new features. Compared to fastp 0.20.0, the new fastp 0.23.2 is even 80% faster.
ImageGP: An easy‐to‐use data visualization web server for scientific researchers
- 21 February 2022
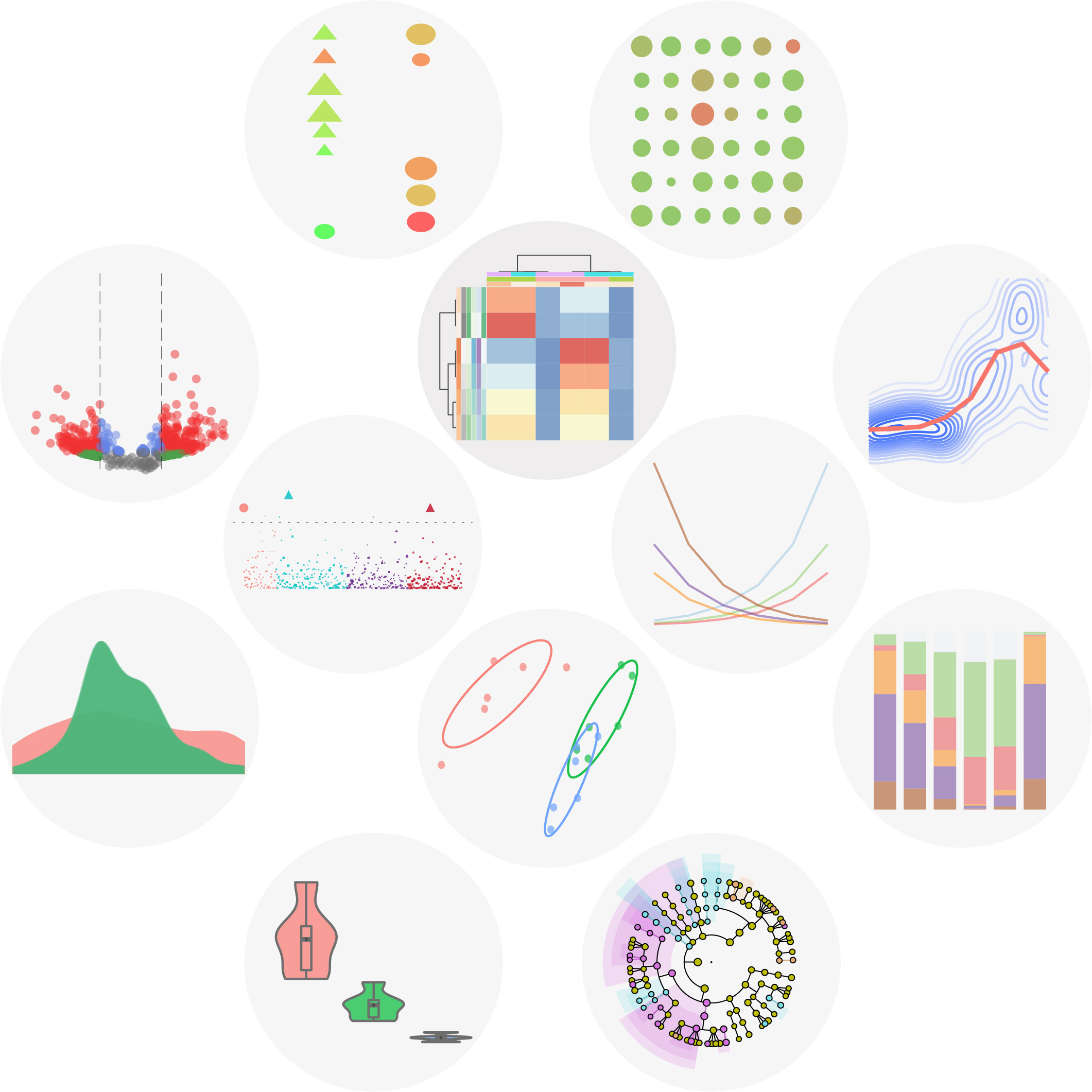
Representative visualization results of ImageGP. ImageGP supports 16 types of images and four types of online analysis with up to 26 parameters for customization. ImageGP also contains specialized plots like volcano plot, functional enrichment plot for most omics-data analysis, and other 4 specialized functions for microbiome analysis. Since 2017, ImageGP has been running for nearly 5 years and serving 336,951 visits from all over the world. Together, ImageGP (http://www.ehbio.com/ImageGP/) is an effective and efficient tool for experimental researchers to comprehensively visualize and interpret data generated from wet-lab and dry-lab.
Ggtree: A serialized data object for visualization of a phylogenetic tree and annotation data
- 28 September 2022
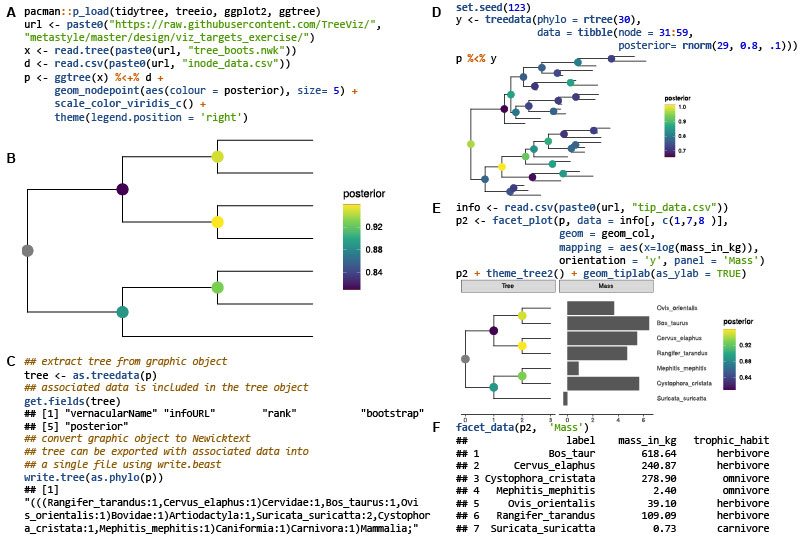
The ggtree object is designed to store phylogenetic tree and associated data, and the object itself is a graphic object that can be rendered as an image file. This work will increase the reproducibility and reusability of phylogenetic data, as well as facilitate integrative and comparative studies.
Using PhyloSuite for molecular phylogeny and tree-based analyses
- 16 February 2023
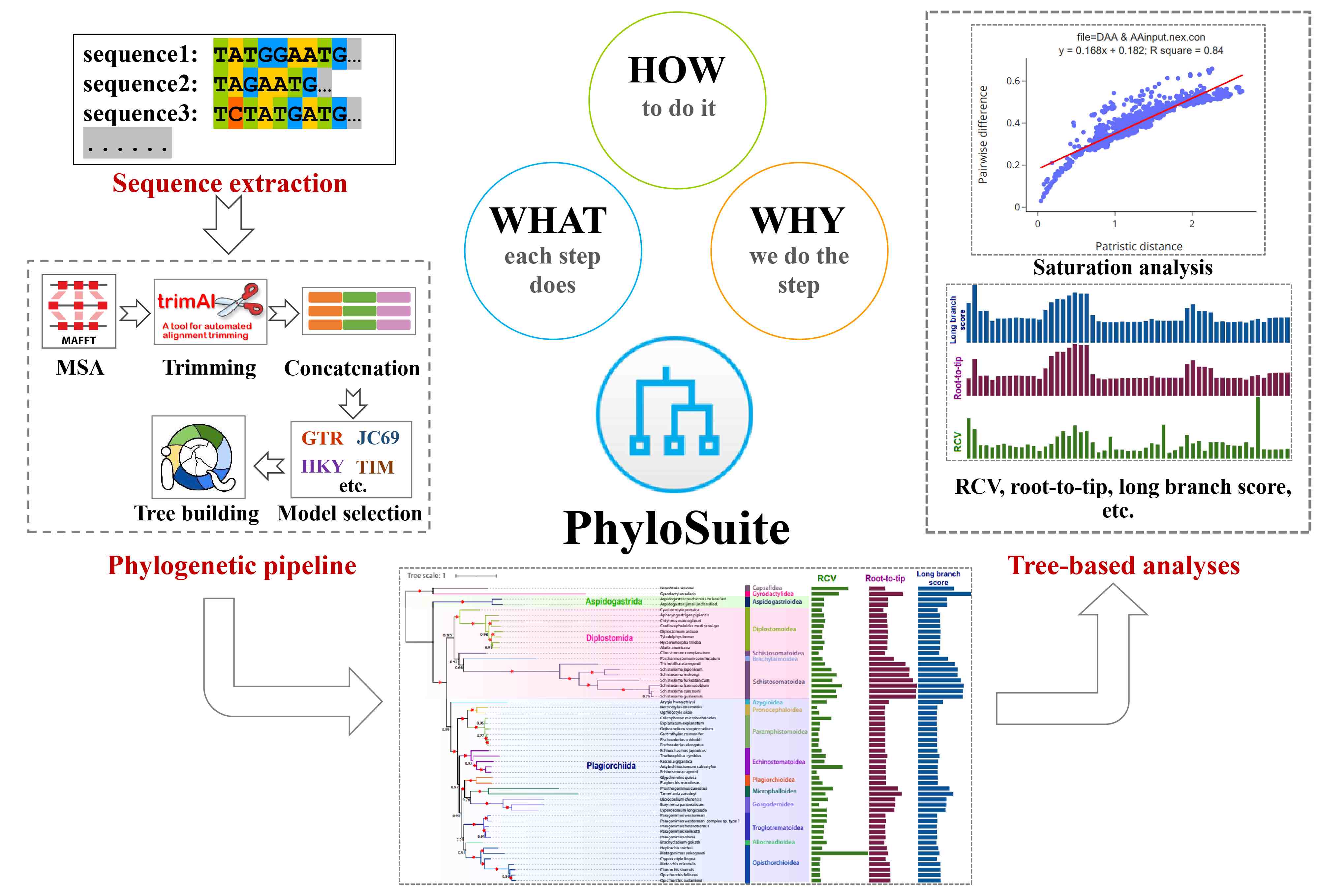
A new release of PhyloSuite, capable of conducting tree-based analyses. Detailed guidelines for each step of phylogenetic and tree-based analyses, following the “What? Why? and How?” structure. This protocol will help beginners learn how to conduct multilocus phylogenetic analyses and help experienced scientists improve their efficiency.
























