
TOmicsVis: An all-in-one transcriptomic analysis and visualization R package with Shinyapp interface
- 13 October 2023
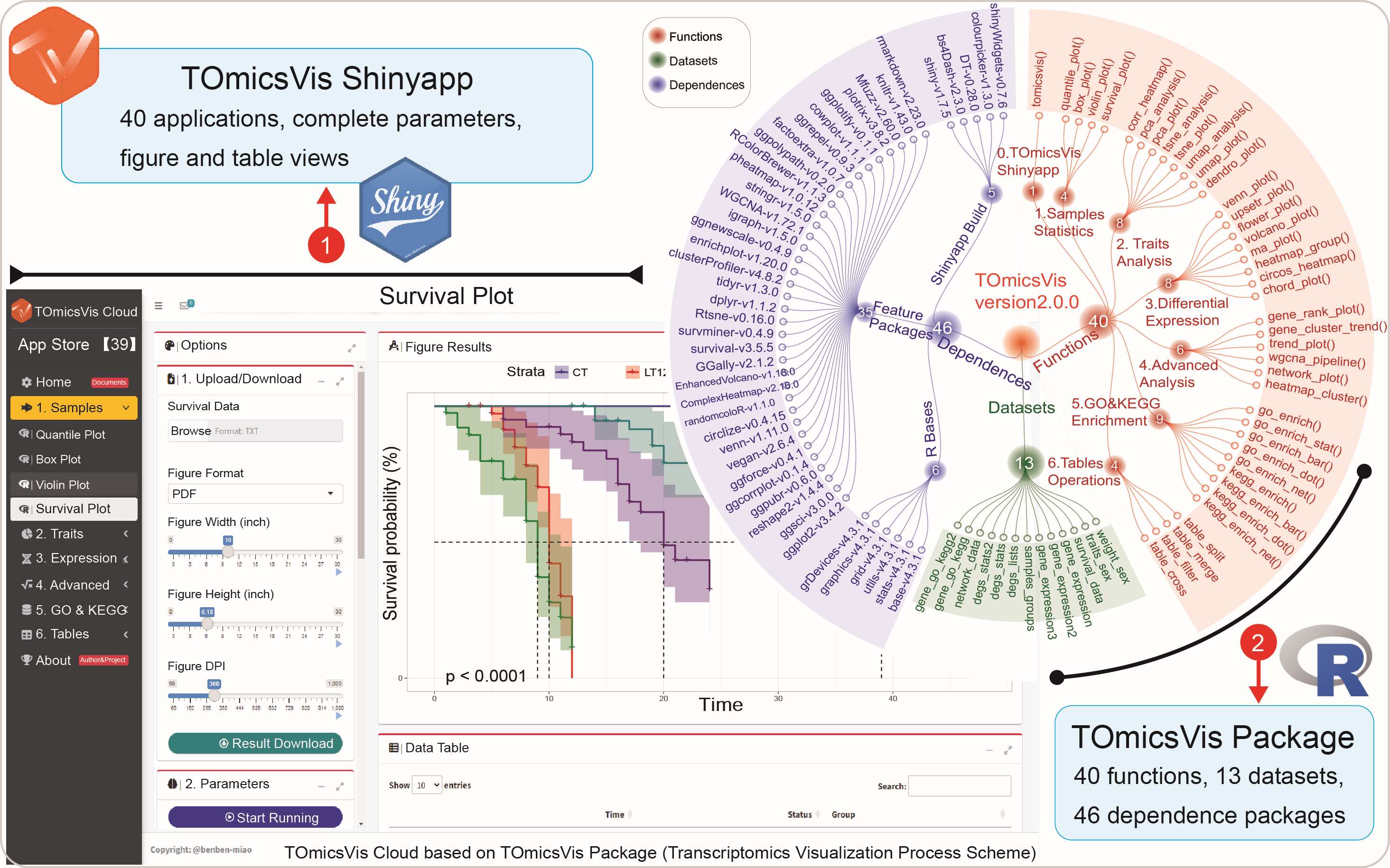
Transcriptomics Visualization (TOmicsVis) implements 40 functions for transcriptomics data analytics and visualization based on 46 R packages, which provide a one-stop analysis and visualization pipeline for two or more groups of transcriptomics projects, with multigrouped example data sets. TOmicsVis offers a local run function for Shinyapp and an online interface analysis service (https://shiny.hiplot.cn/tomicsvis-shiny/), providing significant convenience for researchers without coding skills.
MetaSVs: A pipeline combining long and short reads for analysis and visualization of structural variants in metagenomes
- 12 October 2023
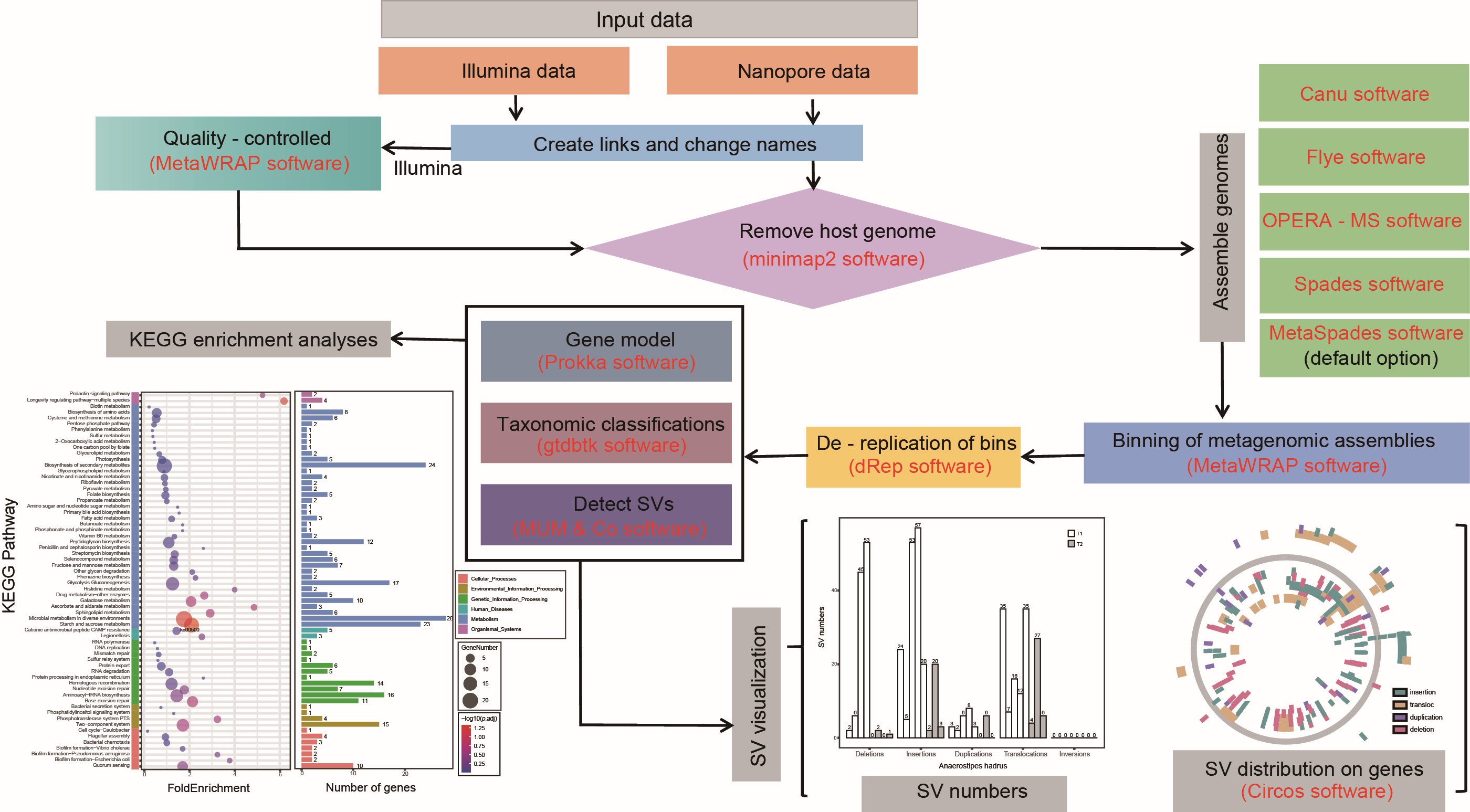
MetaSVs (https://github.com/Wlab518/SV_procedure) are a pipeline combining Nanopore long reads and Illumina short reads to analyze structural variants (SVs) in the microbial genomes and further identify differential SVs that can be reflective of metabolic differences. The pipeline integrates multiple software tools and its core mission consists of 13 steps, including the creation of soft links, quality control, and sequence statistics, removal of host reads, metagenome assembly and evaluation, extraction of high-quality draft genomes (bins), and dereplication, species taxonomy and gene models of bins, detection and visualization of SVs, and Kyoto Encyclopedia of Genes and Genomes enrichment analyses. MetaSVs provide researchers easy access to SVs and relevant metabolites in the microbial genomes without the requirement of specific technical expertise, which is particularly useful to researchers interested in metagenomic SVs but lacking sophisticated bioinformatic knowledge.
ScRNAPip: A systematic and dynamic pipeline for single-cell RNA sequencing analysis
- 31 August 2023
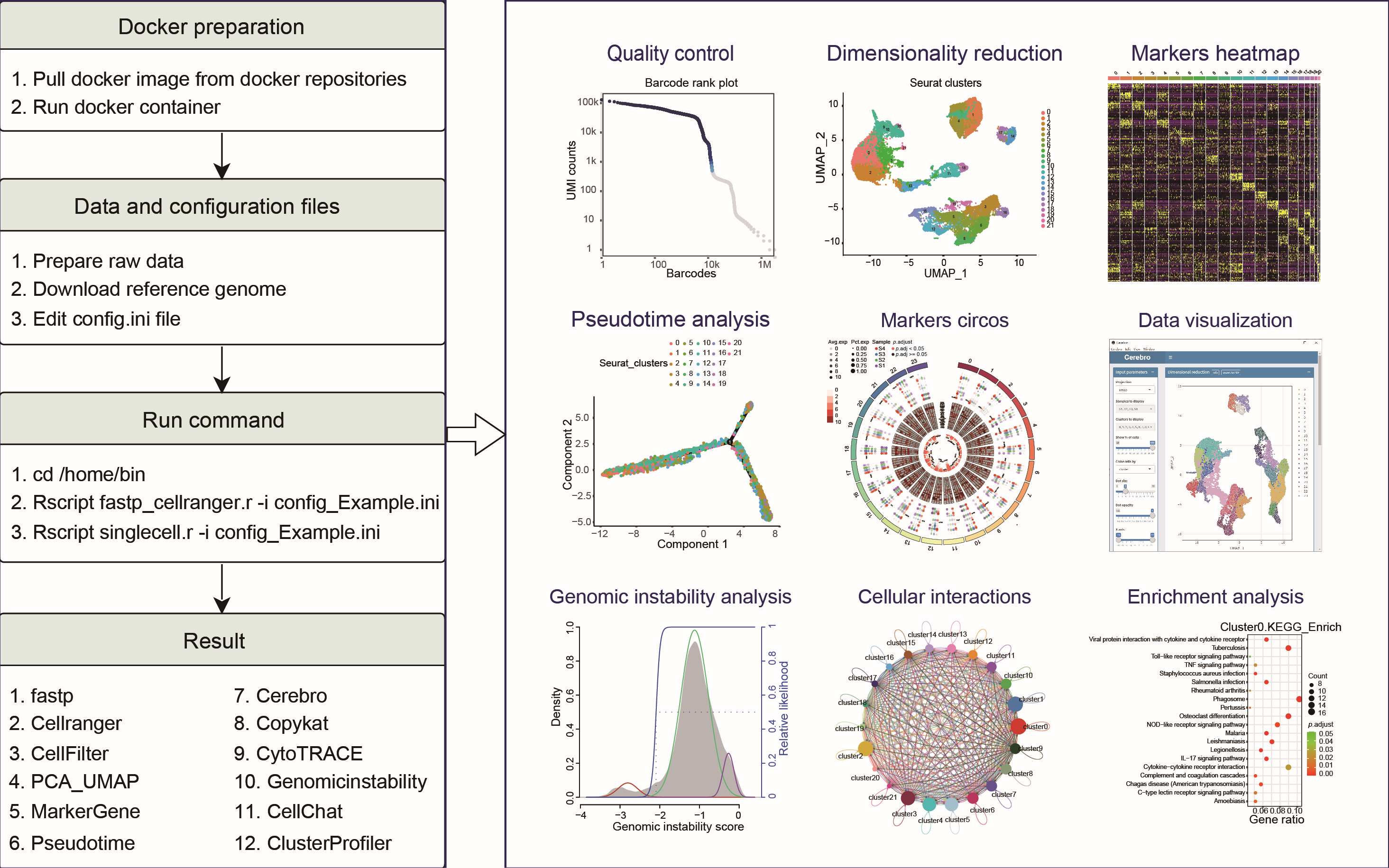
ScRNAPip (https://github.com/OpenGene/scrnapip) is a reproducible computational workflow for scRNA-seq data analysis. It incorporates multiple analysis modules including quality control, normalization, data correction, pseudotime analysis, copy number analysis, cellular interactions, and so on. This study will serve as a tutorial for researchers interested in single-cell data analysis and help users build and update their analysis pipelines.
MultiPrime: A reliable and efficient tool for targeted next-generation sequencing
- 19 October 2023
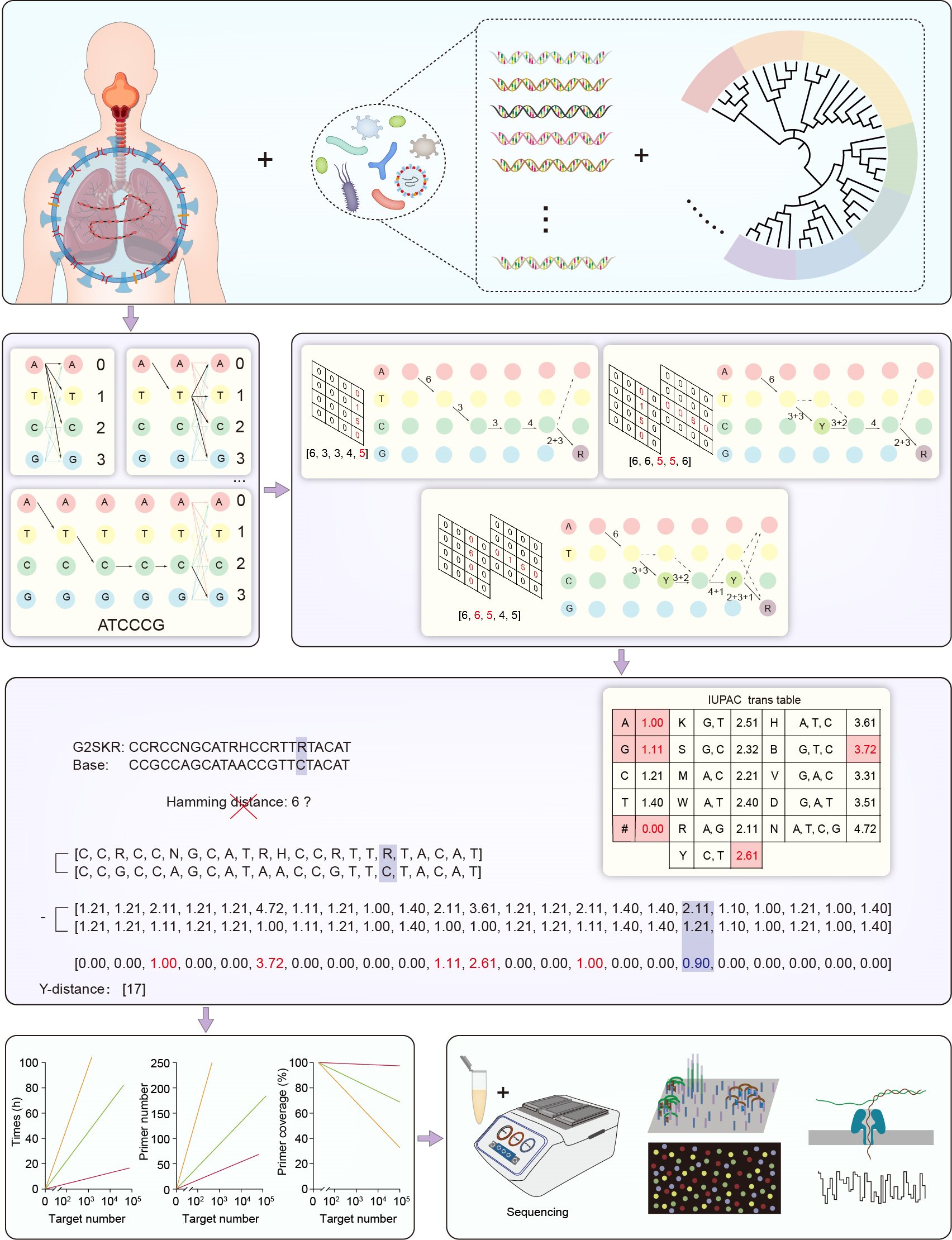
This study introduces multiPrime, a novel tool tailored for the wide-ranging detection of target sequences through targeted next-generation sequencing (tNGS). By integrating degenerate primer design principles with effective mismatch handling, multiPrime demonstrates enhanced precision and specificity in the identification of diverse sequence types. This breakthrough presents a prospective pathway for optimizing the development of tNGS. In performance comparison, multiPrime excelled over conventional tools in terms of execution time, primer coverage, and the number of candidate primers. It offers a streamlined and versatile solution for the rapid and cost-effective detection of diverse microbiomes.
cisDynet: An integrated platform for modeling gene-regulatory dynamics and networks
- 23 November 2023
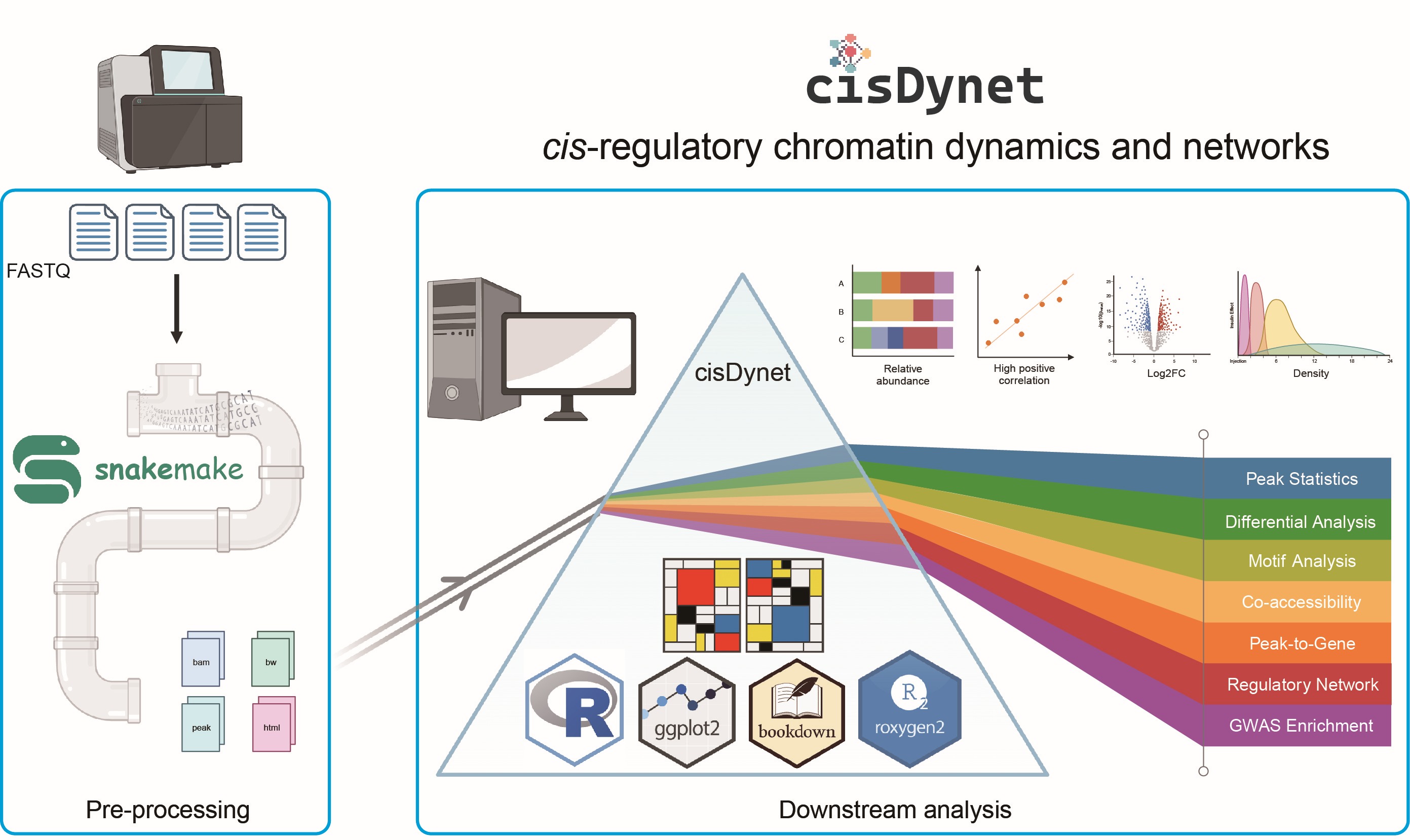
cisDynet enables the exploration of the cis-regulatory chromatin dynamics and networks. It offers a streamlined preprocessing pipeline built on Snakemake, along with an R package for advanced downstream data analysis and visualization. It is free to access on GitHub (https://github.com/tzhu-bio/cisDynet).
Intraspecific difference of Latilactobacillus sakei in inflammatory bowel diseases: Insights into potential mechanisms through comparative genomics and metabolomics analyses
- 25 September 2023
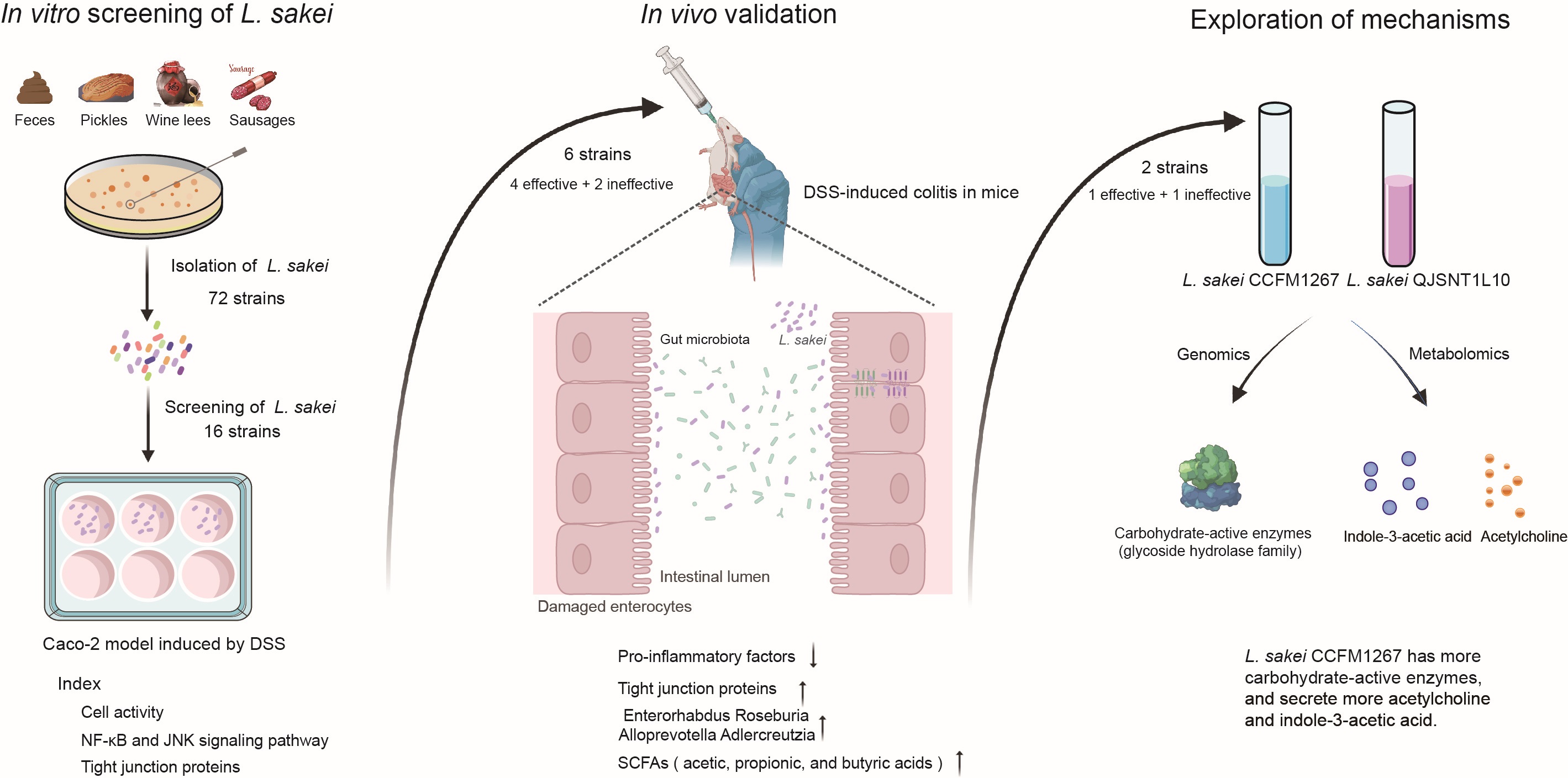
We obtained 72 strains of Latilactobacillus sakei from 120 fermentation and fecal samples across China. In total, 16 strains from different sources were initially screened in an in vitro Caco-2 model. Subsequently, six strains (four exhibiting effectiveness and two exhibiting ineffectiveness) were selected for further validation in an in vivo colitis mouse model. Both in vivo and in vitro findings indicated intraspecific variations of L. sakei in their impacts on inflammatory bowel diseases. Notably, differences in the carbohydrate-active enzymes of L. sakei may exert an indirect influence on the gut microbiota, consequently exerting a more pronounced effect on the short-chain fatty acids, leading to variations in the degree of remission. On the basis of the metabolomic profile of L. sakei strains, it was found that acetylcholine and indole-3-acetic acid were tentatively identified as key substances that may contribute to the variations in their therapeutic efficacy.
Melatonin supplementation protects against traumatic colon injury by regulating SERPINA3N protein expression
- 24 October 2023
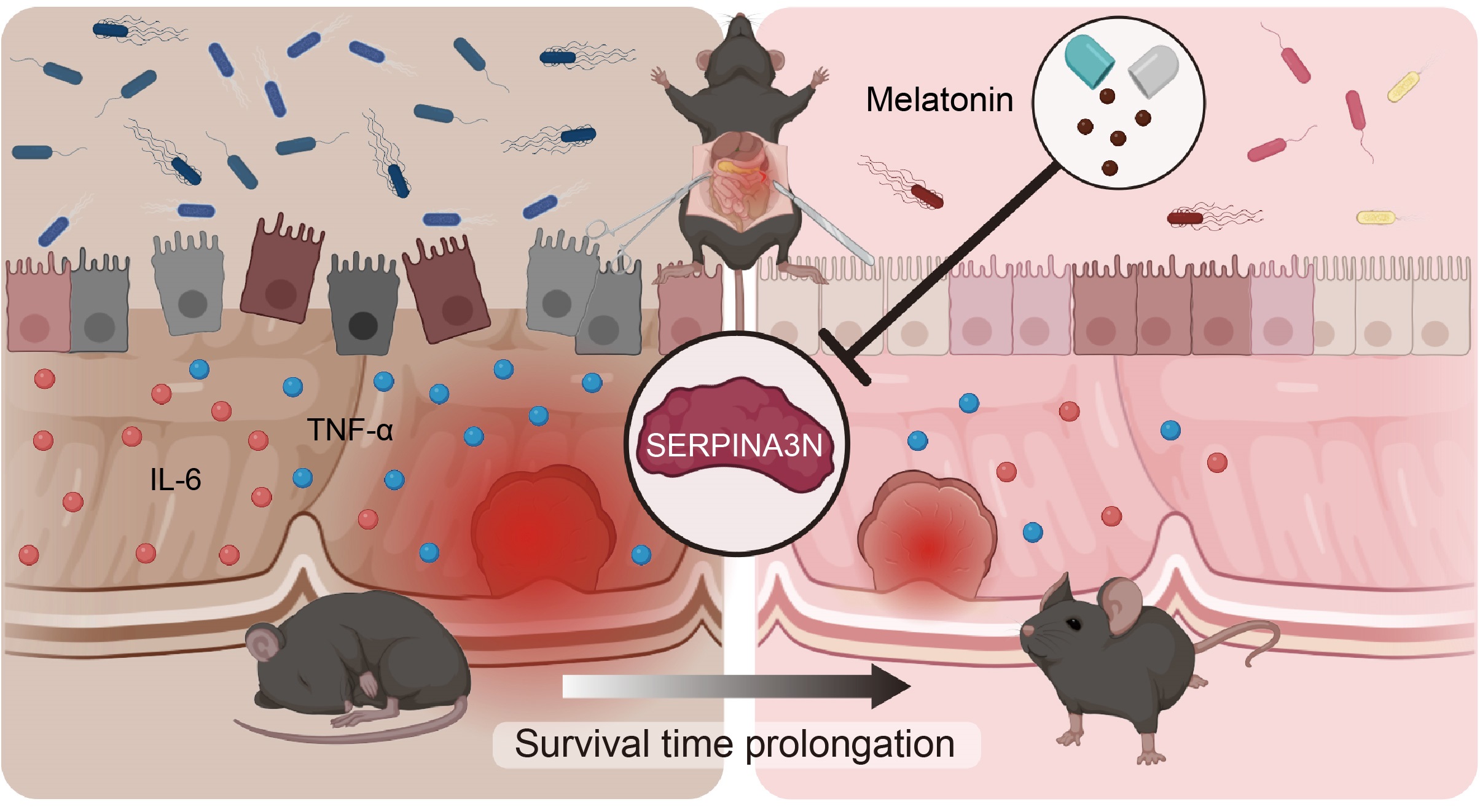
Traumatic colon injury leads to significant disruption of gut homeostasis within a short period of time, indicated by inflammatory responses, intestinal barrier hyperpermeability, and microbiota dysregulation. Melatonin administration can restore gut homeostasis by suppressing SERPINA3N expression in the intestinal epithelium, subsequently prolonging the survival time of mice with traumatic colon injury.
Multiomics characterization and verification of clear cell renal cell carcinoma molecular subtypes to guide precise chemotherapy and immunotherapy
- 16 November 2023
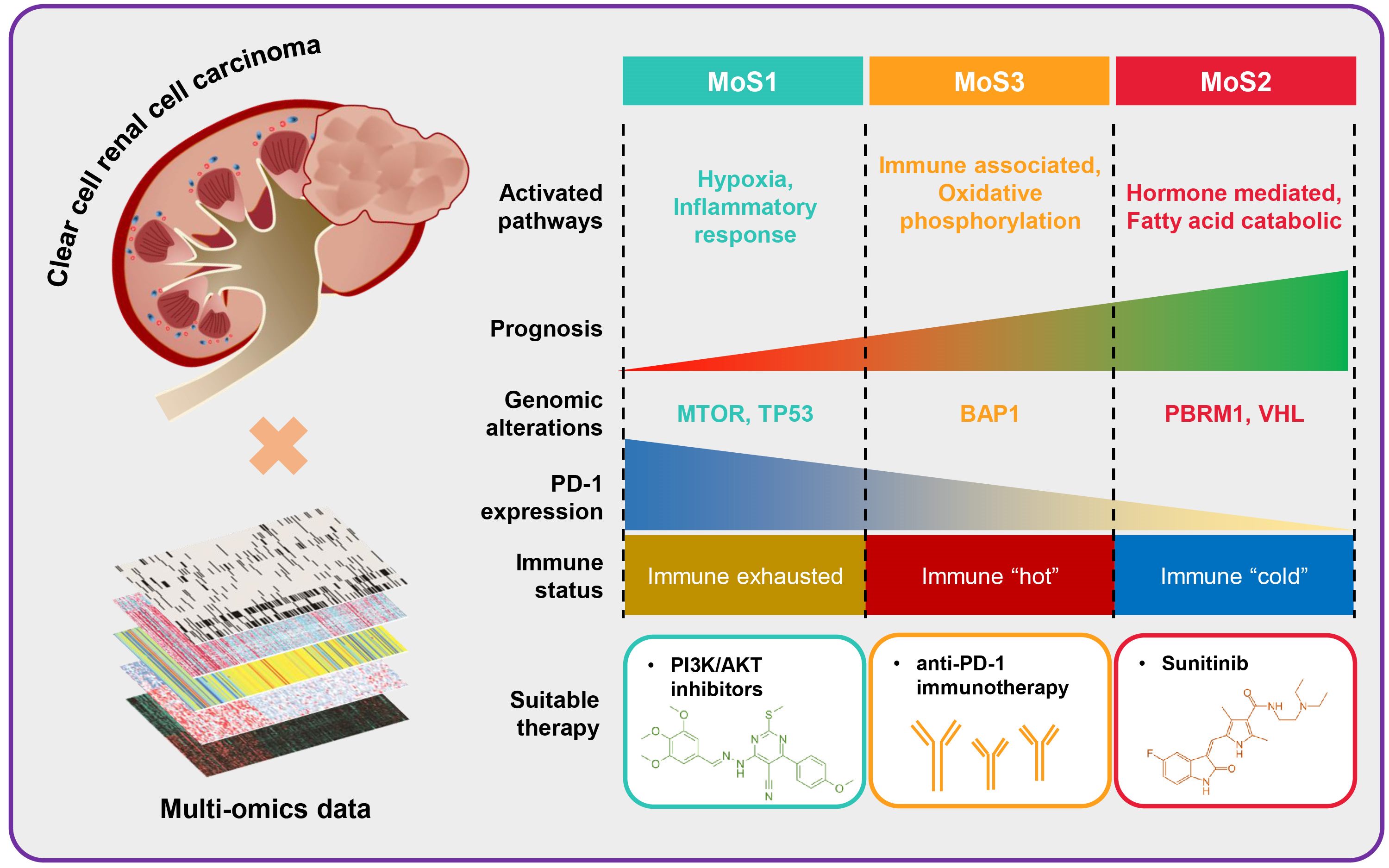
We established a novel molecular classification for ccRCC via multiomics features, named multiomics subtype (MoS). MoS correlates with significantly different clinicopathological parameters, molecular alterations, signaling activation, immunocyte infiltration, and therapeutic response. MoS1 subtype, presented the poorest prognosis, and might be caused by an exhausted immune microenvironment, activated hypoxia features, but can benefit from PI3K/AKT inhibitors. MoS2 subtype represented more mutations of VHL and PBRM1, favorable prognosis and is more suitable for sunitinib therapy. MoS3 is the immune “hot” subtype, and can benefit from the anti-PD-1 immunotherapy. We also confirm that SETD2 is a tumor suppressor in ccRCC, along with the decreased SETD2 protein level in advanced tumor stage, and knock-down of SETD2 leads to the promotion of cell proliferation, migration, and invasion.
Distinct responses of airborne abundant and rare microbial communities to atmospheric changes associated with Chinese New Year
- 11 October 2023
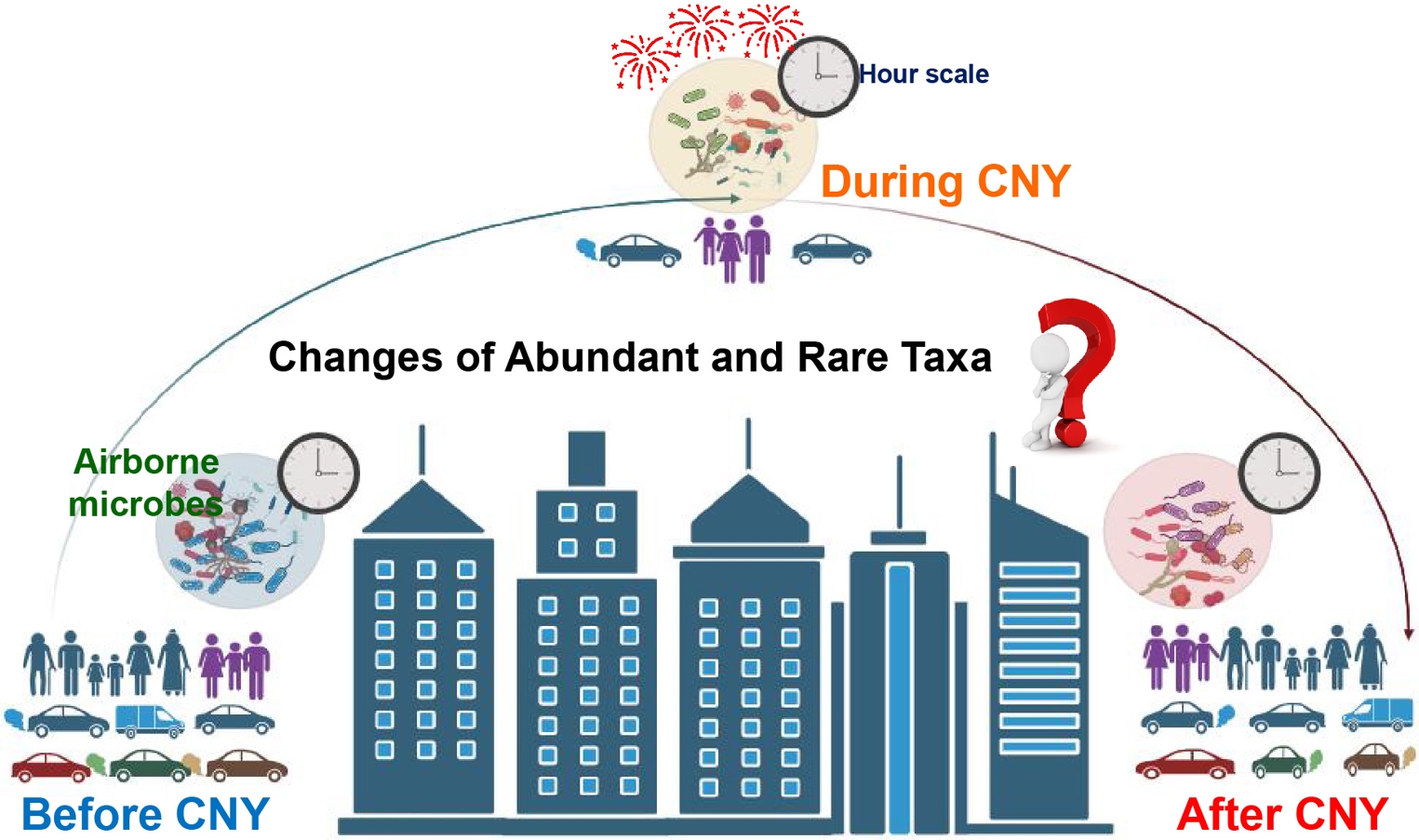
The hour-scale dynamics of bacteria and fungi in the atmosphere were determined. Human activities during Chinese New Year changed the distribution of air pollutants and subsequently affected communities of airborne bacteria and fungi, distinctly. In addition, our results found that abundant species were more sensitive than rare taxa in response to environmental changes associated with Chinese New Year (CNY). The relative abundance of potential bacterial pathogens in the bacterial community was higher during CNY compared with those before and after CNY, which indicated that human activities during CNY increase the threats of airborne microbes to human beings.
Rhizosphere interface microbiome reassembly by arbuscular mycorrhizal fungi weakens cadmium migration dynamics
- 31 August 2023
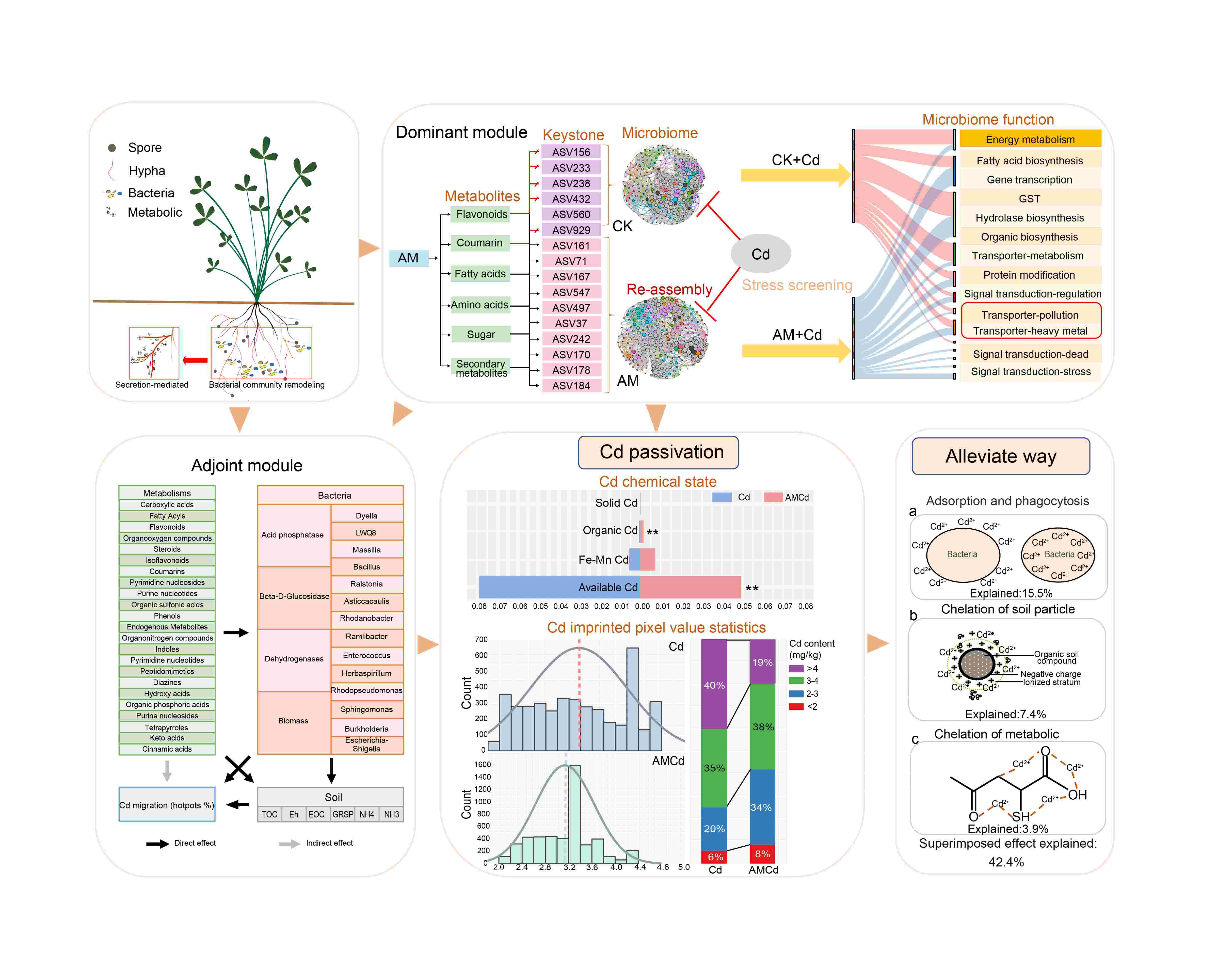
Arbuscular mycorrhizal fungi (AMF) reduced Cd migration in the rhizosphere through microbial community variation, exhibiting a distinct deterministic process, which ultimately resulted in a core microbiome function of heavy metal resistance and nutrient cycling. The above process is related to upregulating root metabolites by AMF, with flavonoids, coumarin, fatty acids and so on being the main factor causing microbiome variation. Additionally, the superposition of the root metabolites, microbial, and soil make contributed to Cd migration reduction, and the microbial model had the highest single explanation rate. Thus, the AMF in the rhizosphere microenvironment can regulate metabolite-soil-microbial interactions, reducing Cd migration.
From heart to gut: Exploring the gut microbiome in congenital heart disease
- 30 October 2023
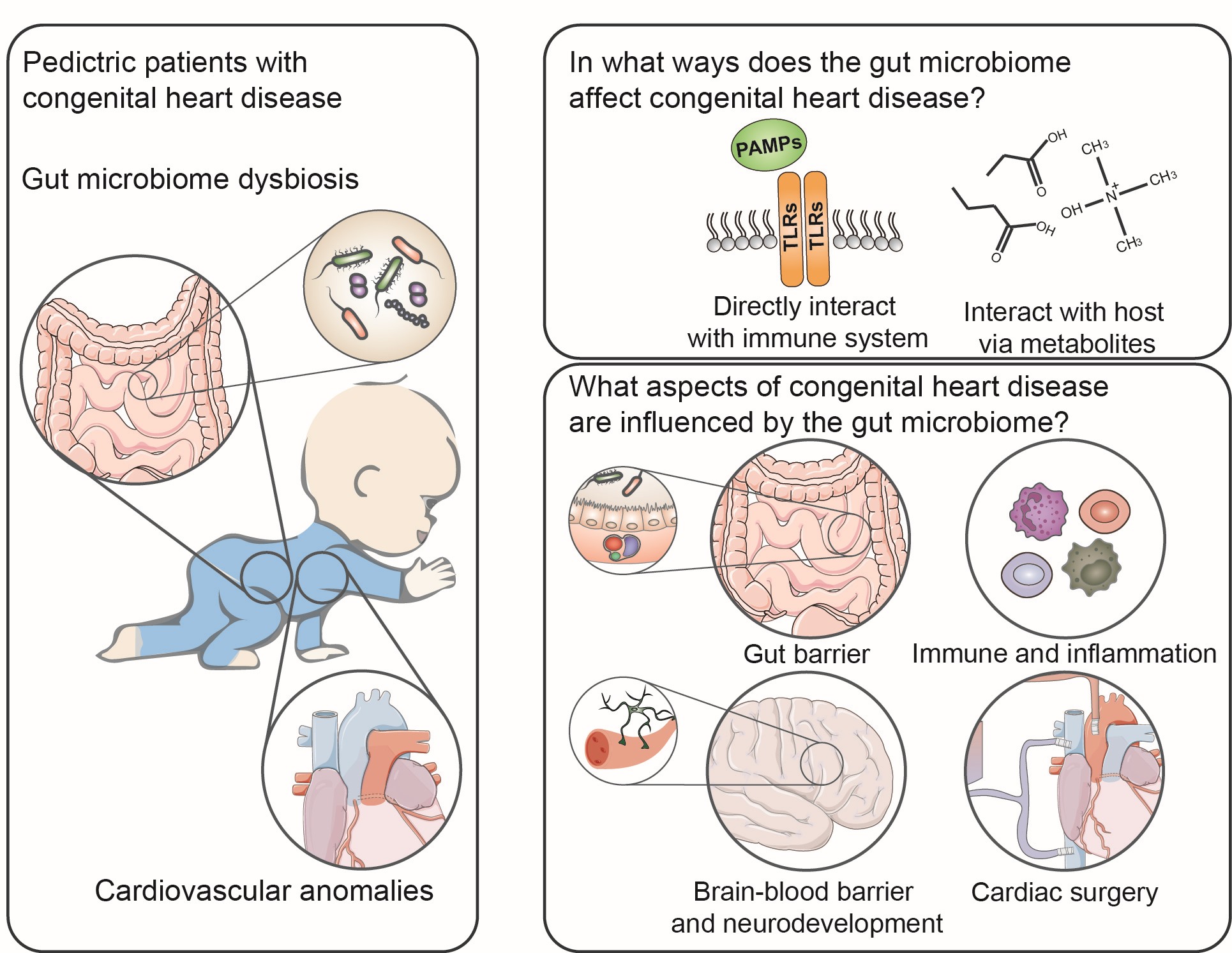
Children with congenital heart disease (CHD) experience gut microbiome dysbiosis in their early life. The gut microbiome affects CHD via immune stimulation and microbiome metabolites, influencing immunity, gut barrier, and inflammation. The bidirectional interaction between the gut microbiome and CHD plays an important role in CHD's development, treatment, and prognosis.
VeloPro: A pipeline integrating Ribo-seq and AlphaFold deciphers association patterns between translation velocity and protein structure features
- 19 November 2023
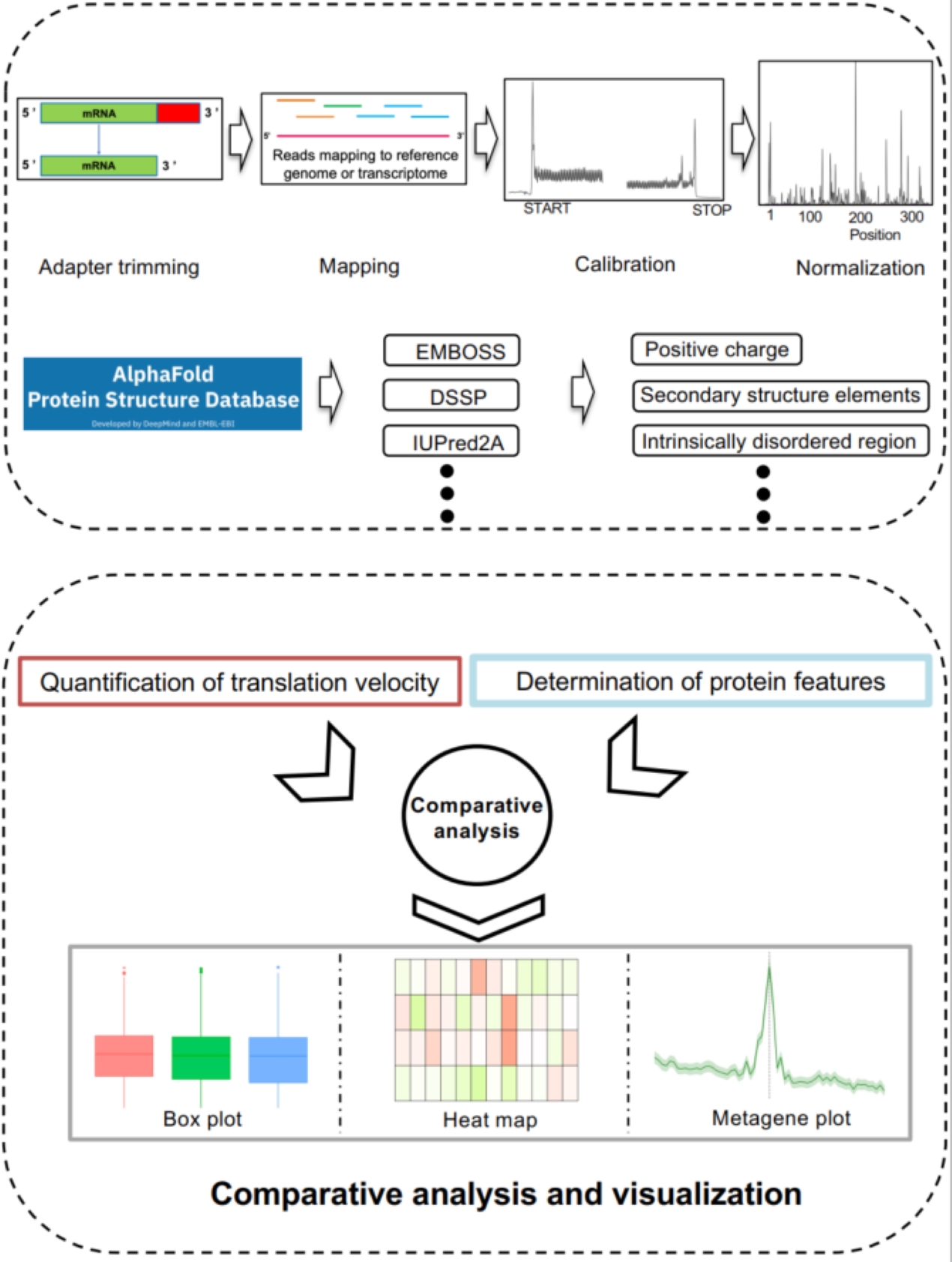
VeloPro integrates Ribo-seq data and AlphaFold2-predicted 3D protein structure information for characterization of the association patterns between translation velocity and many protein structure features in prokaryotic and eukaryotic organisms across different taxonomical clades such as bacteria, fungi, protozoa, nematode, plants, insect, and mammals. We illustrated that association patterns between translation velocity and protein structure features differ across organisms, partially reflecting their taxonomical relationship.
MIST: A microbial identification and source tracking system for next-generation sequencing data
- 02 November 2023
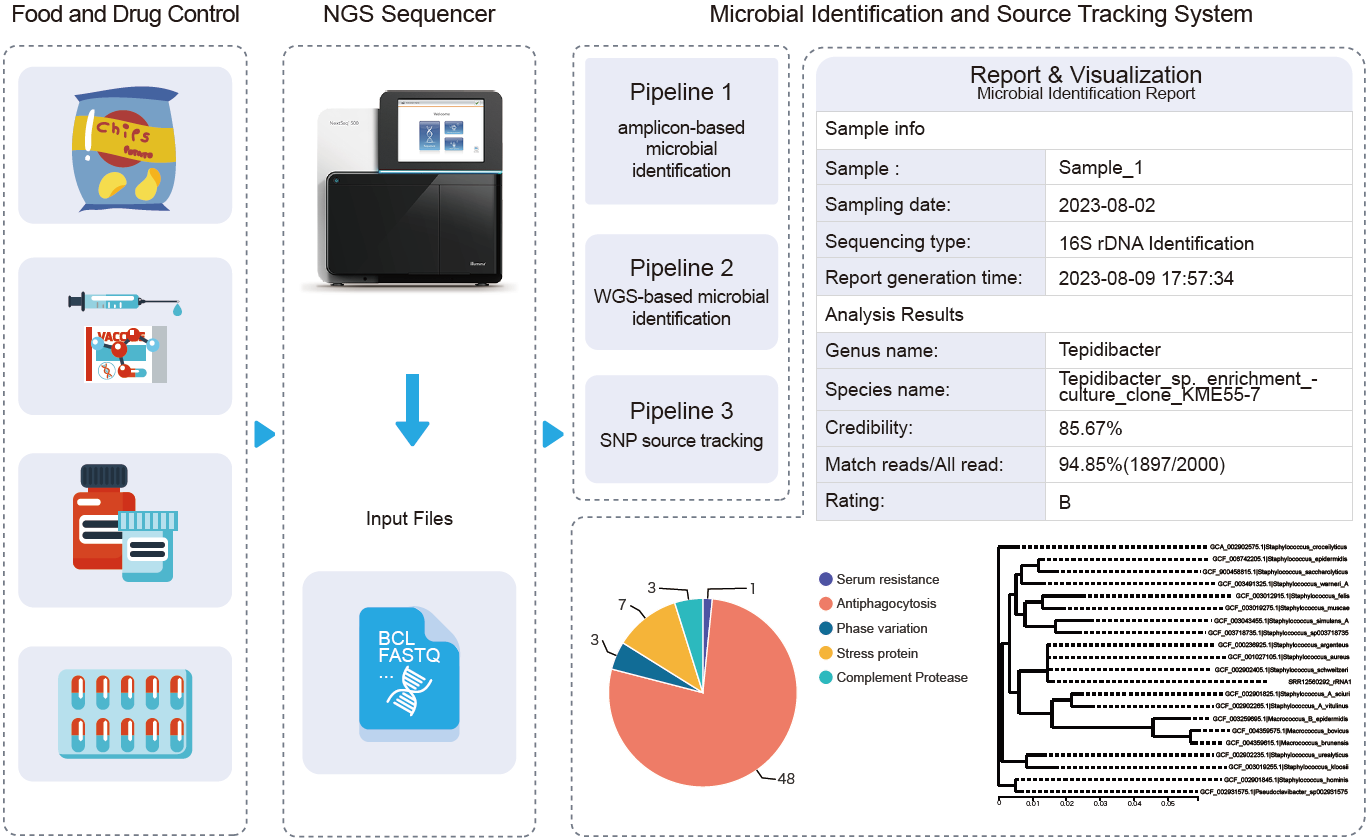
The Professional Committee of Microbiology of the National Pharmacopoeia Commission organized the drafting of the Technical Guidelines for Microbial Whole Genome Sequencing (WGS), aiming to standardize the method process and technical indicators of microbial WGS and ensure the accuracy of sequencing and identification. On the basis of the Guidelines, we developed an integrated microbial identification and source tracking (MIST) system, which could meet the needs of microbial identification and contamination investigation in food and drug quality control. MIST integrates three analysis pipelines: 16S/18S/internal transcribed spacer amplicon-based microbial identification, WGS-based microbial identification, and single-nucleotide polymorphism-based microbial source tracking. MIST can analyze sequence data in a variety of formats, such as Fasta, base call file, and FASTQ. It can be connected to a high-throughput sequencing instrument to acquire sequencing data directly. We also developed a publicly accessible web server for MIST (http://syj.i-sanger.cn).
Primer selection impacts the evaluation of microecological patterns in environmental microbiomes
- 17 September 2023
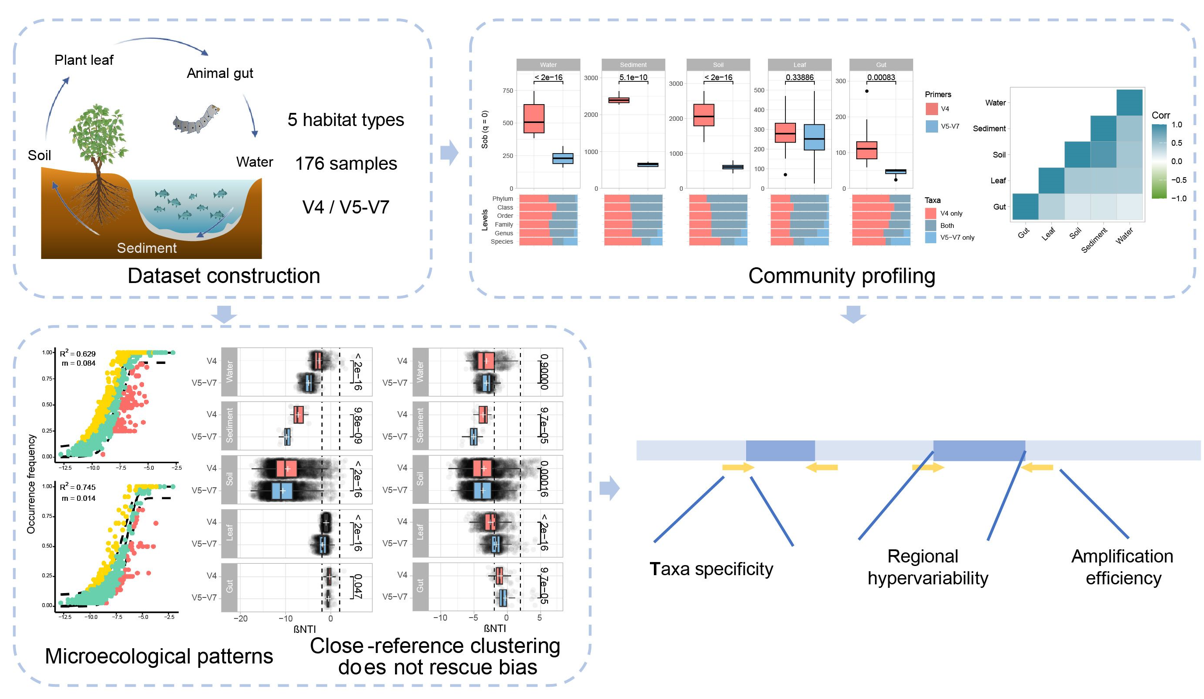
This study revealed that primer selection substantially influences the taxonomic and predicted functional composition and the characterization of microecological patterns, which was not alleviated by close-reference clustering. Biases were relatively consistent across different habitats in community profiling but not in microecological patterns. These primer biases could be attributed to multiple aspects, including taxa specificity, regional hypervariability, and amplification efficiency.
MicroEXPERT: Microbiome profiling platform with cross-study metagenome-wide association analysis functionality
- 17 August 2023
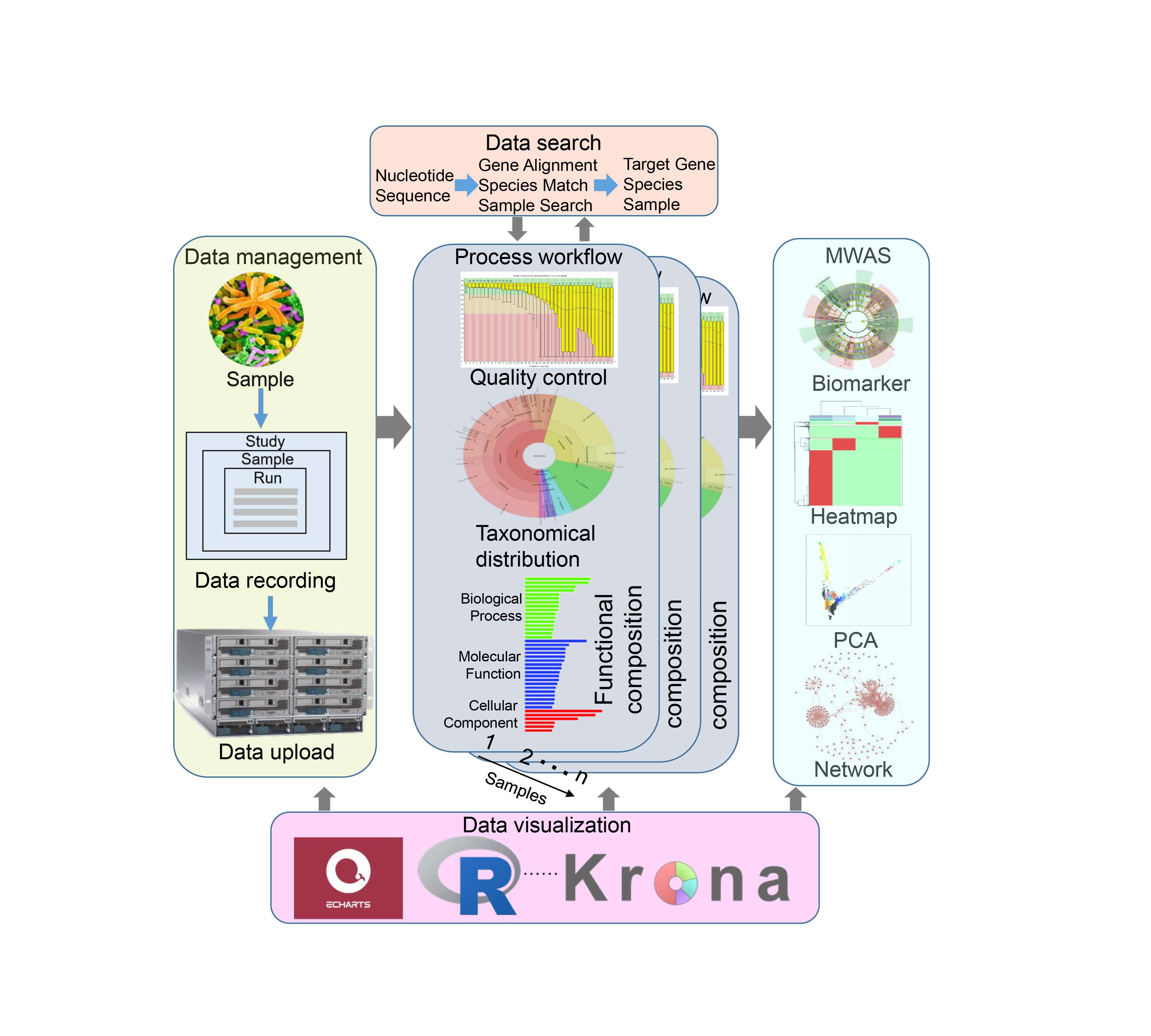
The framework of the MicroEXPERT platform. Our Platform was composed of five modules. Data management module: Users upload raw data and metadata to the system using a guided workflow. Data processing module: Uploaded data is processed to generate taxonomical distribution and functional composition results. Metagenome-wide association studies module (MWAS): Various methods, including biomarker analysis, PCA, co-occurrence networks, and sample classification, are employed using metadata. Data search module: Users can query nucleotide sequences to retrieve information in the MicroEXPERT database. Data visualization module: Visualization tools are used to illustrate the metagenome analysis results.
Host selection tendency of key microbiota in arid desert lichen crusts
- 25 September 2023
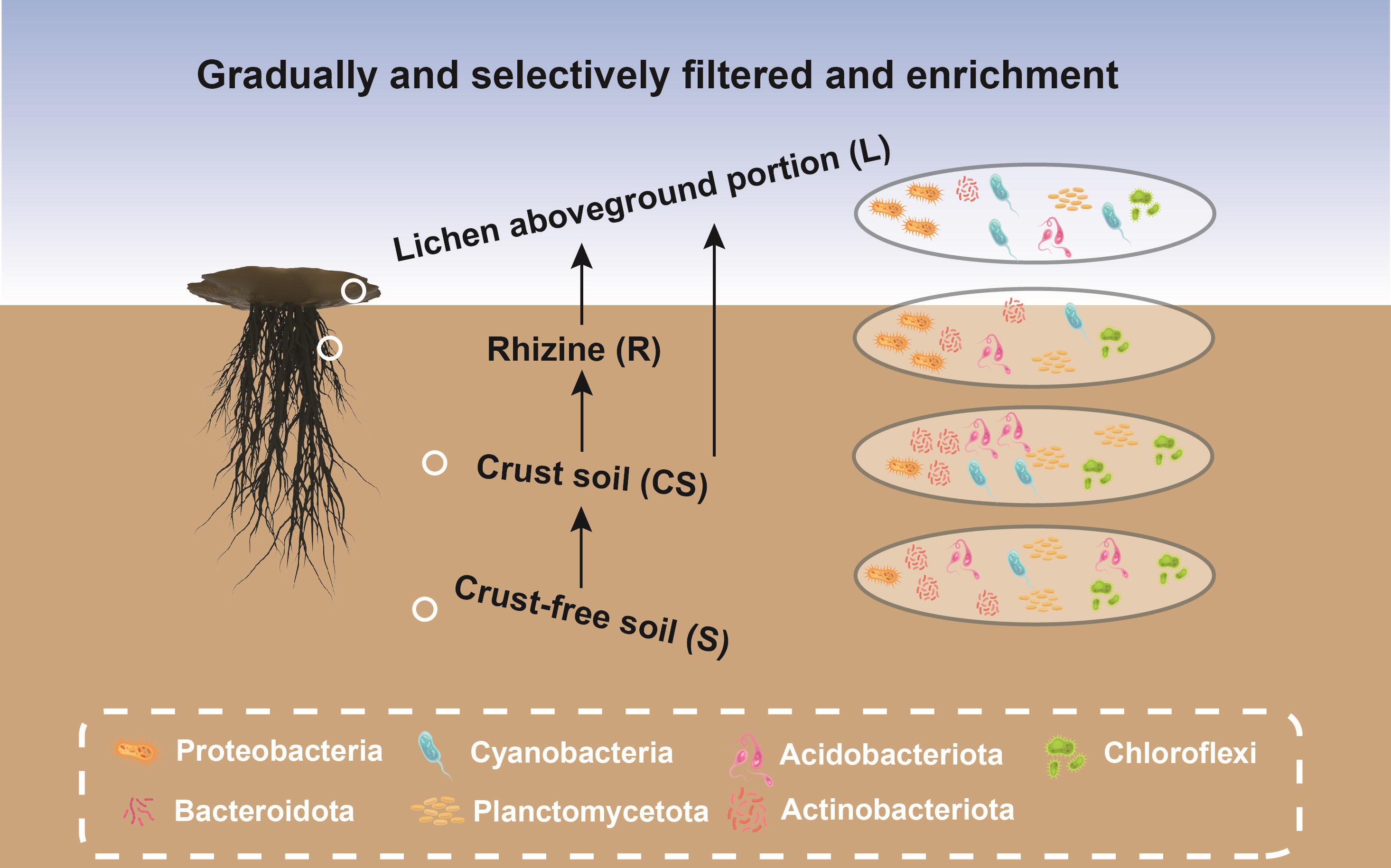
Lichen genus Endocarpon in biological soil crust form was chosen as a model to investigate the bacterial communities for the first time across four vertically distinct strata. Key bacterial microbiota in lichen thallus were discovered, which were gradually filtered and mainly derived from the crust soil, with clear host selection tendency. The study provided key information to better understand the homeostasis maintenance mechanism of the lichen symbiont and community assembly of desert lichen crust.
Soil labile organic carbon fractions mediate microbial community assembly processes during long-term vegetation succession in a semiarid region
- 22 October 2023
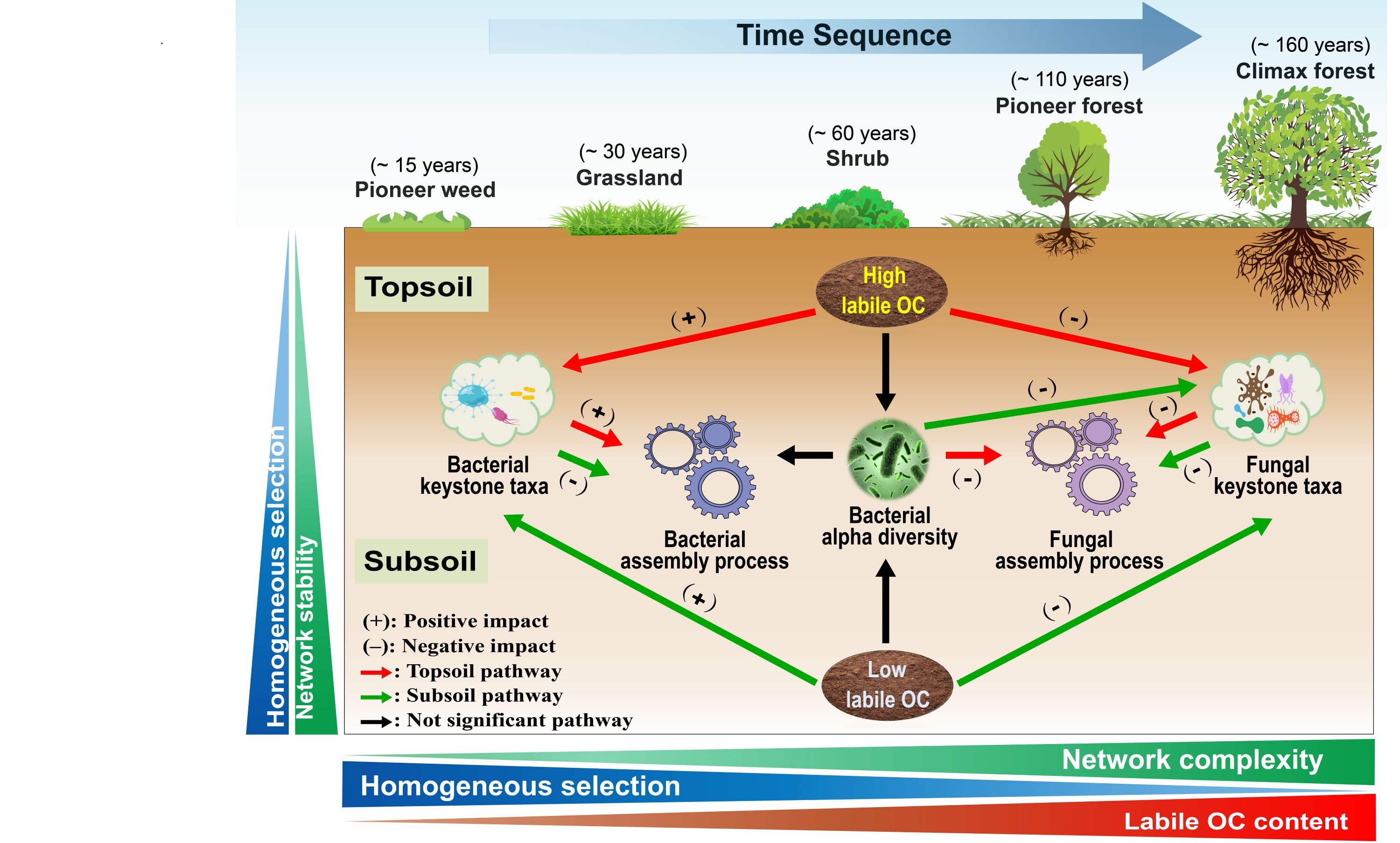
Conceptual diagram for the labile organic carbon (OC) fractions mediating microbial assembly processes during long-term vegetation succession.
Wheat straw hydrochar induced negative priming effect on carbon decomposition in a coastal soil
- 13 September 2023
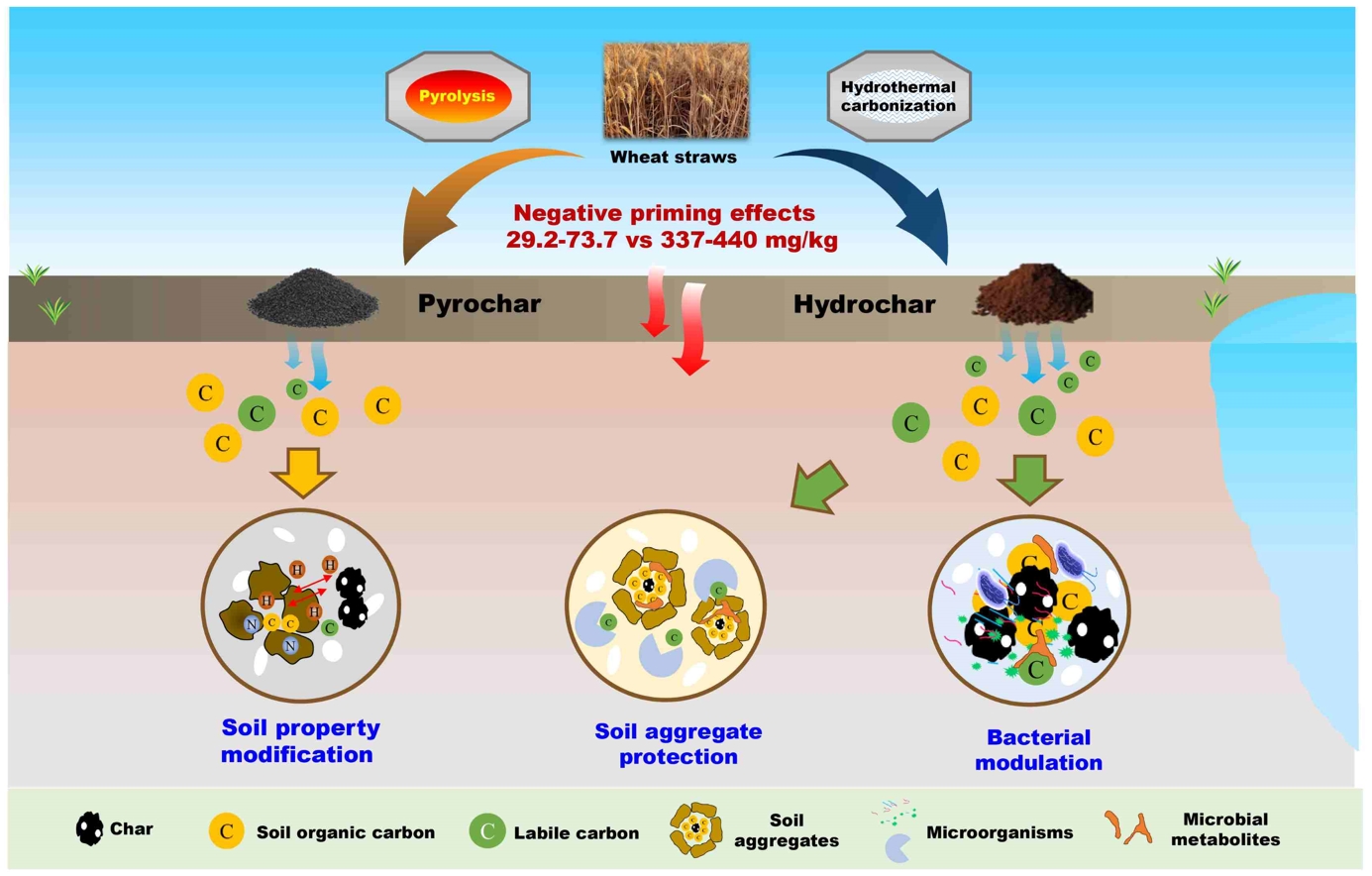
The mechanisms underlying hydrochar-regulated soil organic carbon (SOC) decomposition in the coastal salt-affected soils were first investigated. Straw-derived hydrochar (SHC)-induced C-transformation bacterial modulation and soil aggregation enhancement primarily accounted for negative priming effects. Modification of soil properties (e.g., decreased pH and increased C/N ratios) by straw-derived pyrochar (SPC) was responsible for decreased SOC decomposition.









