
Ggtree: A serialized data object for visualization of a phylogenetic tree and annotation data
- 28 September 2022
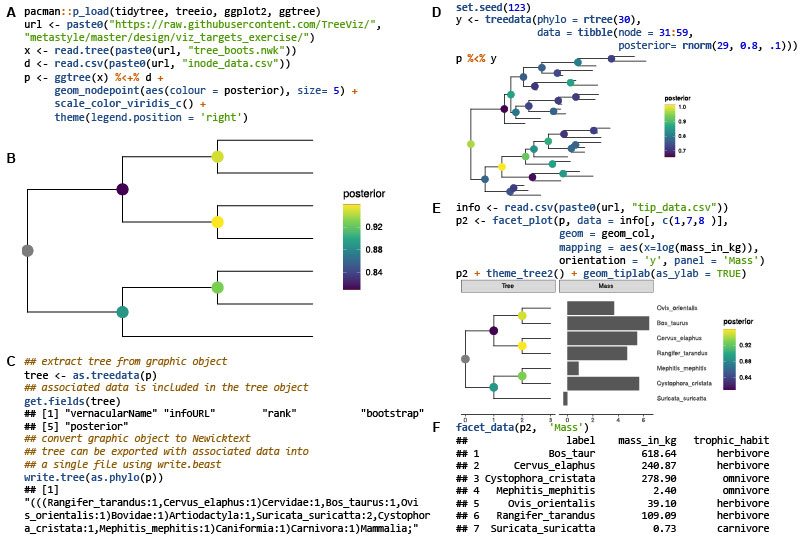
The ggtree object is designed to store phylogenetic tree and associated data, and the object itself is a graphic object that can be rendered as an image file. This work will increase the reproducibility and reusability of phylogenetic data, as well as facilitate integrative and comparative studies.
IPGA: A handy integrated prokaryotes genome and pan-genome analysis web service
- 14 September 2022

Integrated Prokaryotes Genome and pan-genome Analysis (IPGA) serves as a free and easy-to-use web-based system that could provide up-to-date pan-genome analysis service for non-bioinformaticians. IPGA offers users the most reliable pan-genome profile which enables users to perform additional comparative genomic analysis. IPGA provides a series of downstream analysis modules such as phylogenetic inference, synteny inference, and target genome annotation.
TCM-Suite: A comprehensive and holistic platform for Traditional Chinese Medicine component identification and network pharmacology analysis
- 15 August 2022
TCM-Suite platform is composed of two sub-databases, Holmes-Suite and Watson-Suite. Holmes-Suite database covers six marker genes, resulting in 235,470 kinds of biological ingredients. Watson-Suite retrieved massive data for mining the “herb-compound-protein-disease” interpretation. A holistic pipeline was designed to connect Holmes and Watson, showed by a user-friendly platform.
TCM2COVID: A resource of anti-COVID-19 traditional Chinese medicine with effects and mechanisms
- 05 August 2022
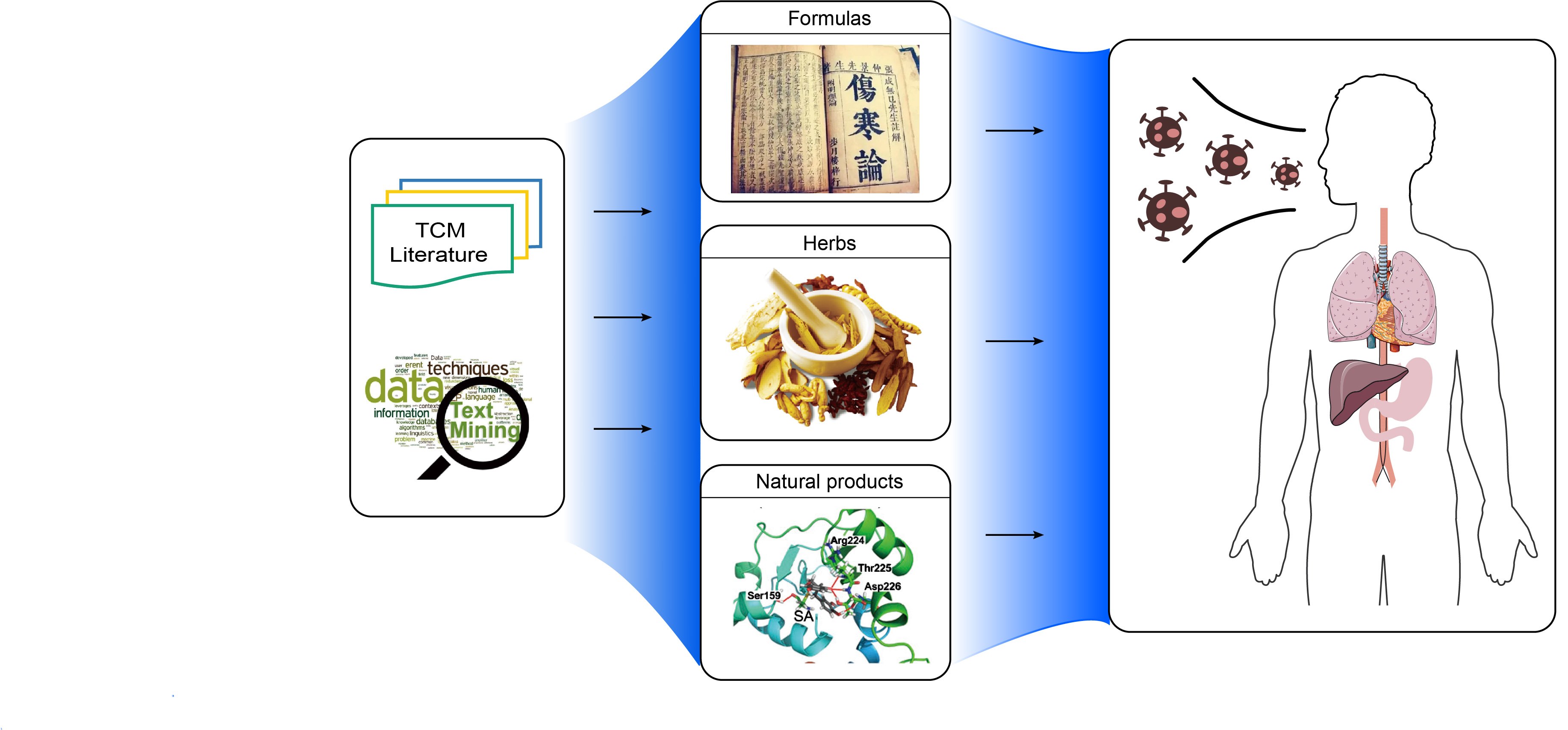
TCM2COVID documents over 280 traditional Chinese medicine formulas (including over 300 herbs) with detailed clinical evidence and therapeutic mechanism information; TCM2COVID records over 80 natural products with detailed potential therapeutic mechanisms; TCM2COVID launches a useful web server for querying, analyzing, and visualizing documented formulas similar to those supplied by the user (formula similarity analysis).
mibPOPdb: An online database for microbial biodegradation of persistent organic pollutants
- 17 August 2022
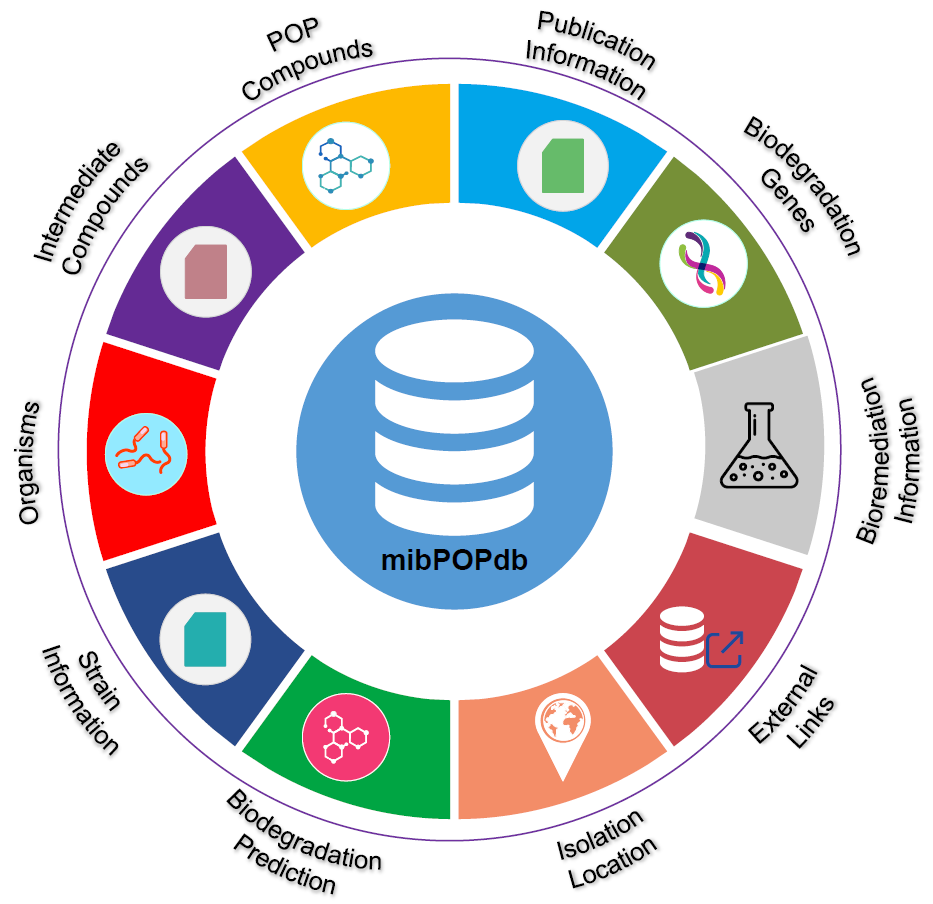
This study presents the Microbial Biodegradation of Persistent Organic Pollutants Database (mibPOPdb) database, a web-accessible literature-based microbial biodegradation of persistent organic pollutants (POPs) resource. We also developed a robust chemical biodegradability prediction model with Graph Neural Networks. By providing high-level curated information on POP-degrading microbial communities, mibPOPdb will be an essential platform for fostering studies on microbial biodegradation of POP compounds and how these microbes would help solve the problem of POP accumulation. In addition, the in silico model can be used to evaluate the persistence of organic chemicals, which is a critical task in ecological risk assessment studies.
MetaTrass: A high-quality metagenome assembler of the human gut microbiome by cobarcoding sequencing reads
- 15 August 2022
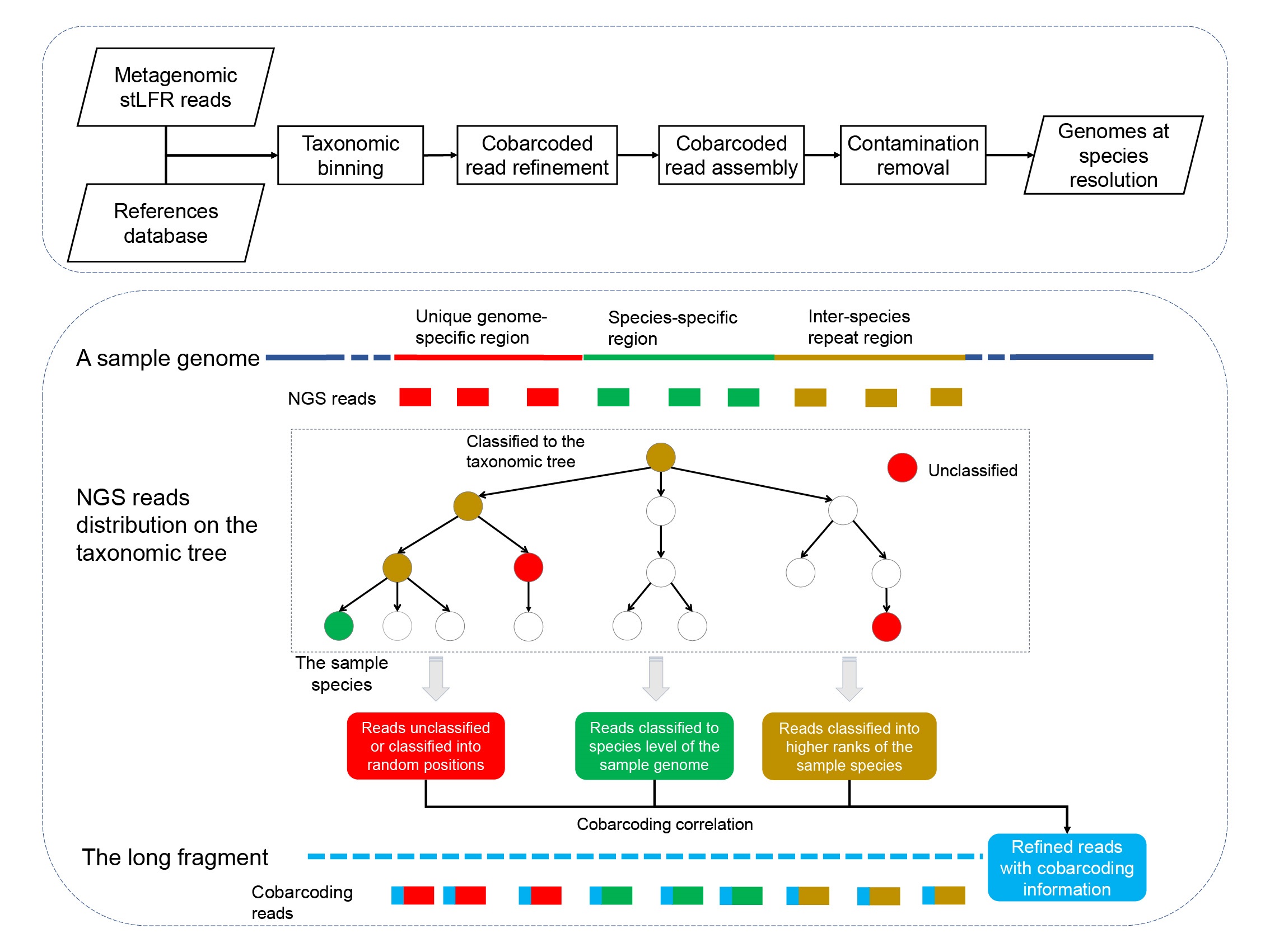
We developed a tool to get high-quality genomes with high taxonomic resolution by combining the cobarcoding information with public references. Compared with the conventional combination strategies, our pipeline generated a large number of high-quality genomes for the human microbial cobarcoding data sets.
Metabolite profiling of human-originated Lachnospiraceae at the strain level
- 13 October 2022
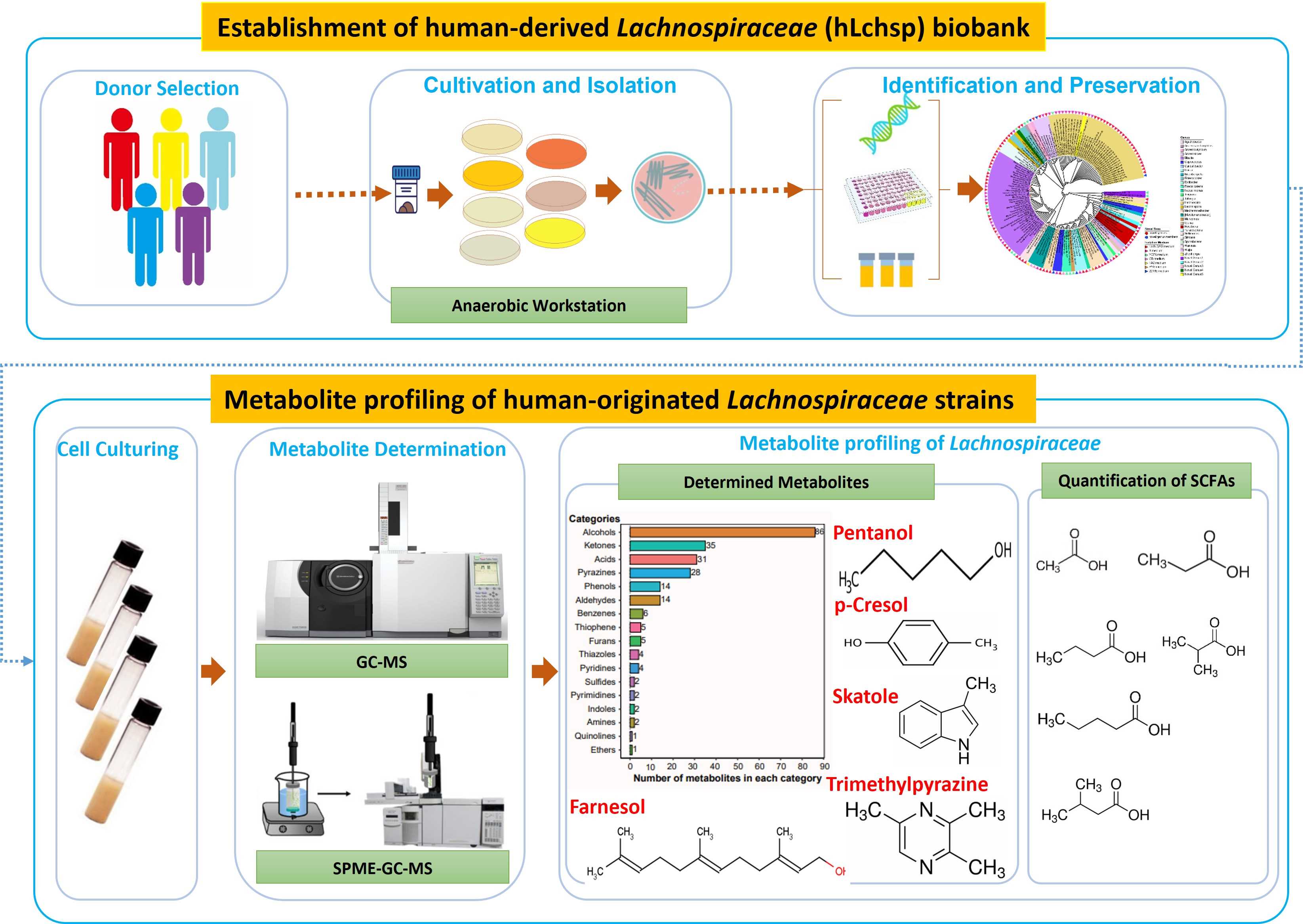
The human-originated Lachnospiraceae biobank included 77 species was constructed. In vitro metabolite profiling of 110 Lachnospiraceae strains yielded 242 metabolites of 17 categories. Many Lachnospiraceae strains produce short-chain fatty acids, and Agathobacter rectalis strain Lach-101 and Coprococcus comes strain NSJ-173 are the top two butyric acid producers in vitro.
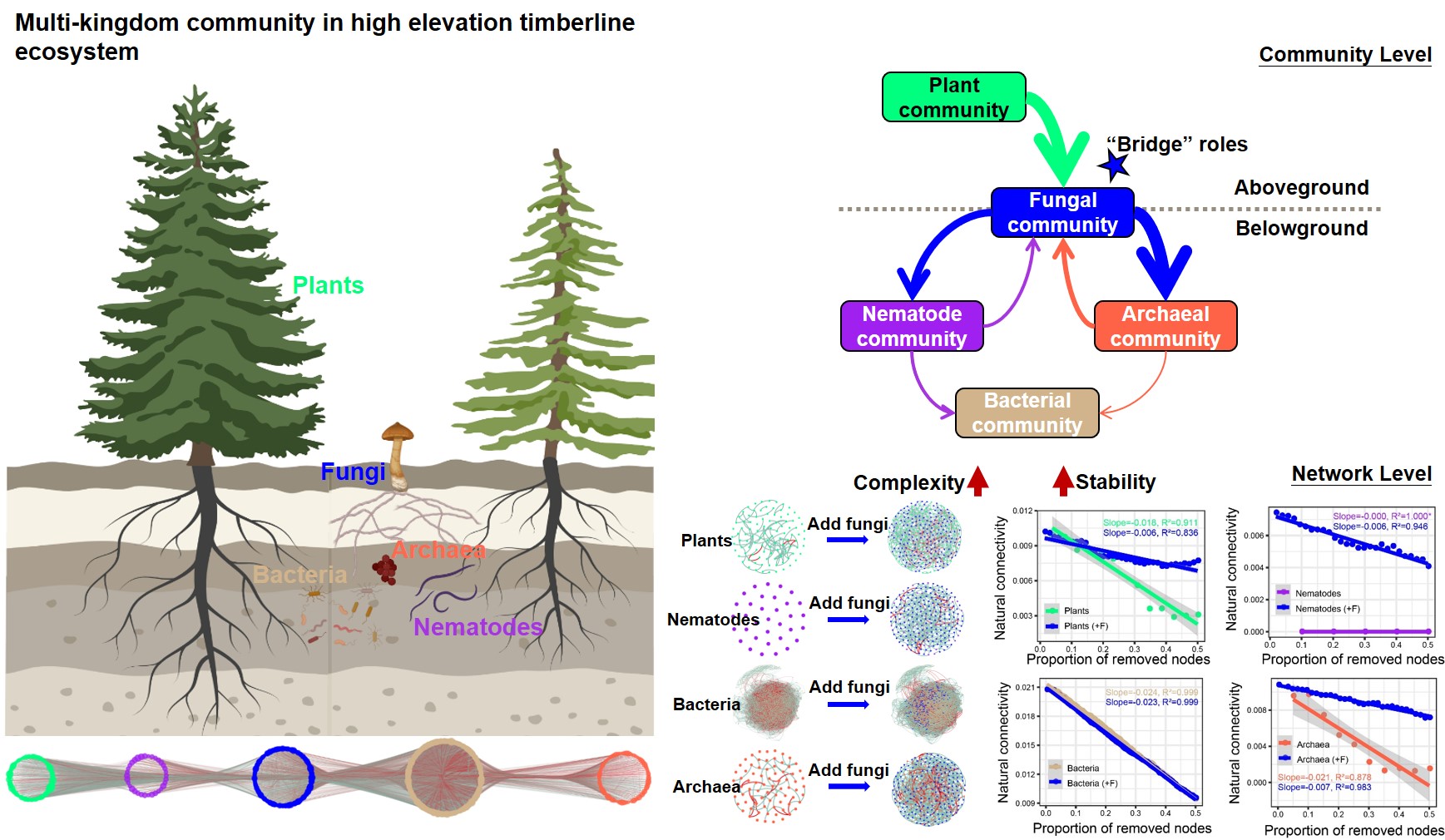
Fungi stabilize multi-kingdom community in a high elevation timberline ecosystem
- 15 August 2022
TCM-Suite platform is composed of two sub-databases, Holmes-Suite and Watson-Suite. Holmes-Suite database covers six marker genes, resulting in 235,470 kinds of biological ingredients. Watson-Suite retrieved massive data for mining the “herb-compound-protein-disease” interpretation. A holistic pipeline was designed to connect Holmes and Watson, showed by a user-friendly platform.
Gut microbiota composition in the sympatric and diet-sharing Drosophila simulans and Dicranocephalus wallichii bowringi shaped largely by community assembly processes rather than regional species pool
- 13 October 2022
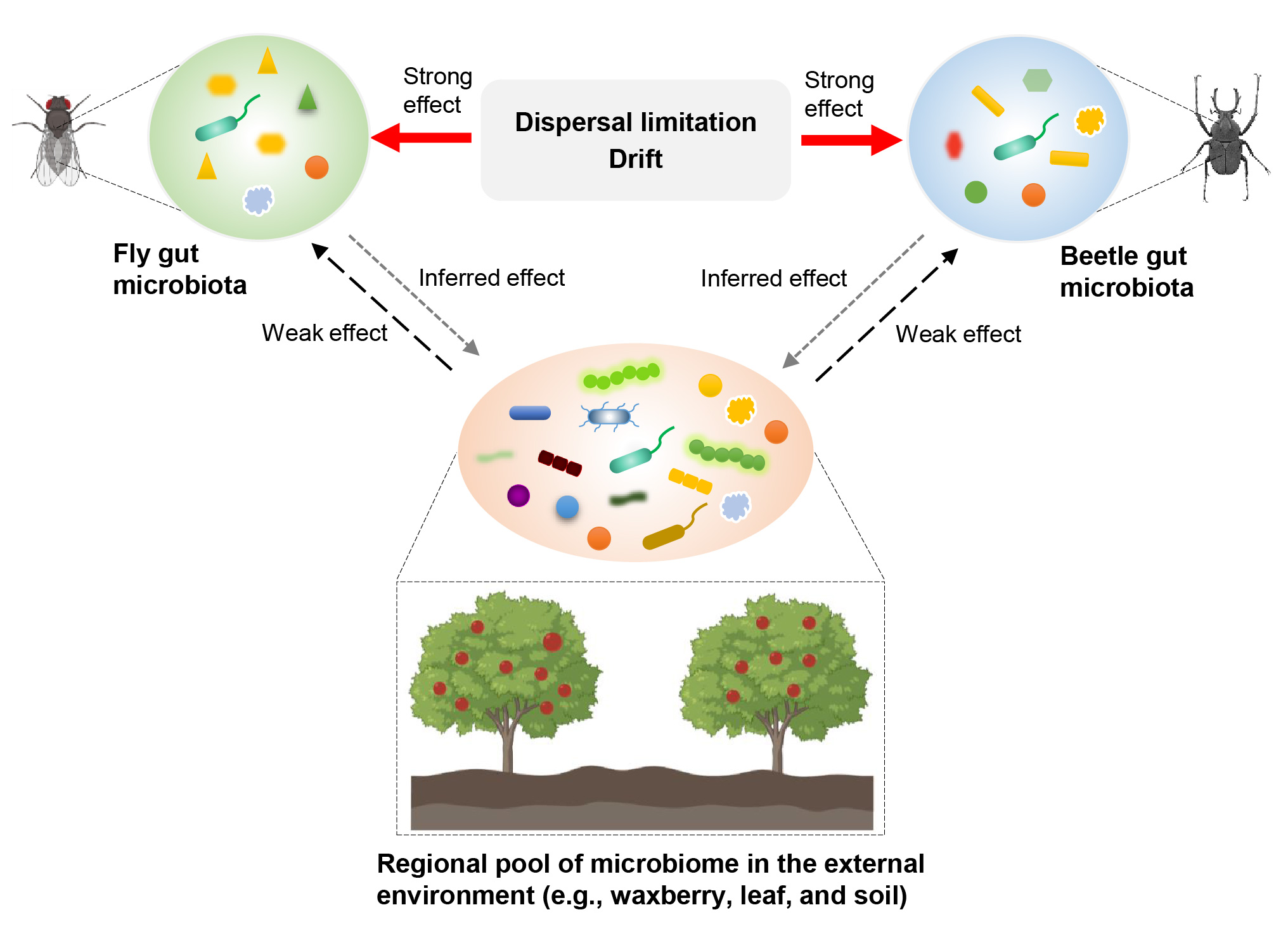
The diversity, composition, and network of gut microbiota differed between the sympatric and diet-sharing Drosophila simulans and Dicranocephalus wallichii bowringi. Host species shape the bacterial and fungal community in two insect hosts by altering the relative contribution of community assembly processes. A minority of gut microbiota within D. simulans and D. wallichii bowringi are drawn from a regional microbial pool from waxberries, leaves, or soil. The composition of insect gut microbiota is driven by community assembly processes in a host species-dependent manner more than regional microbial pools.
Targeting keystone species helps restore the dysbiosis of butyrate-producing bacteria in non-alcoholic fatty liver disease
- 16 November 2022
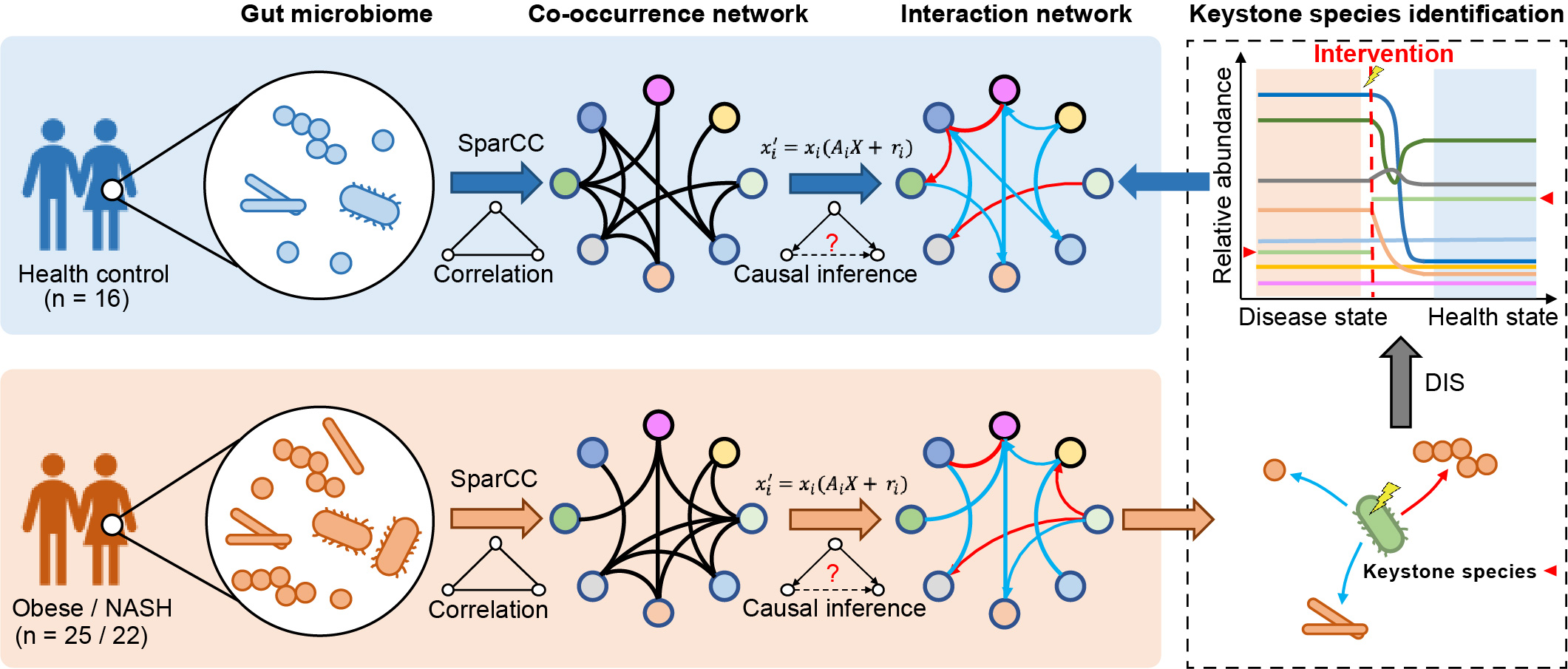
In this study, we applied an algorithm to the keystone species identification in the gut microbiome, based on current causal inference theories and the dynamic intervention simulation. We identified the nonalcoholic steatohepatitis (NASH) keystone species combination, represented by Porphyromonas loveana, Alistipes indistinctus, and Dialister pneumosintes, that showed the highest potential for the microbial intervention of NASH.
Dietary fiber chemical structure determined gut microbiota dynamics
- 28 November 2022
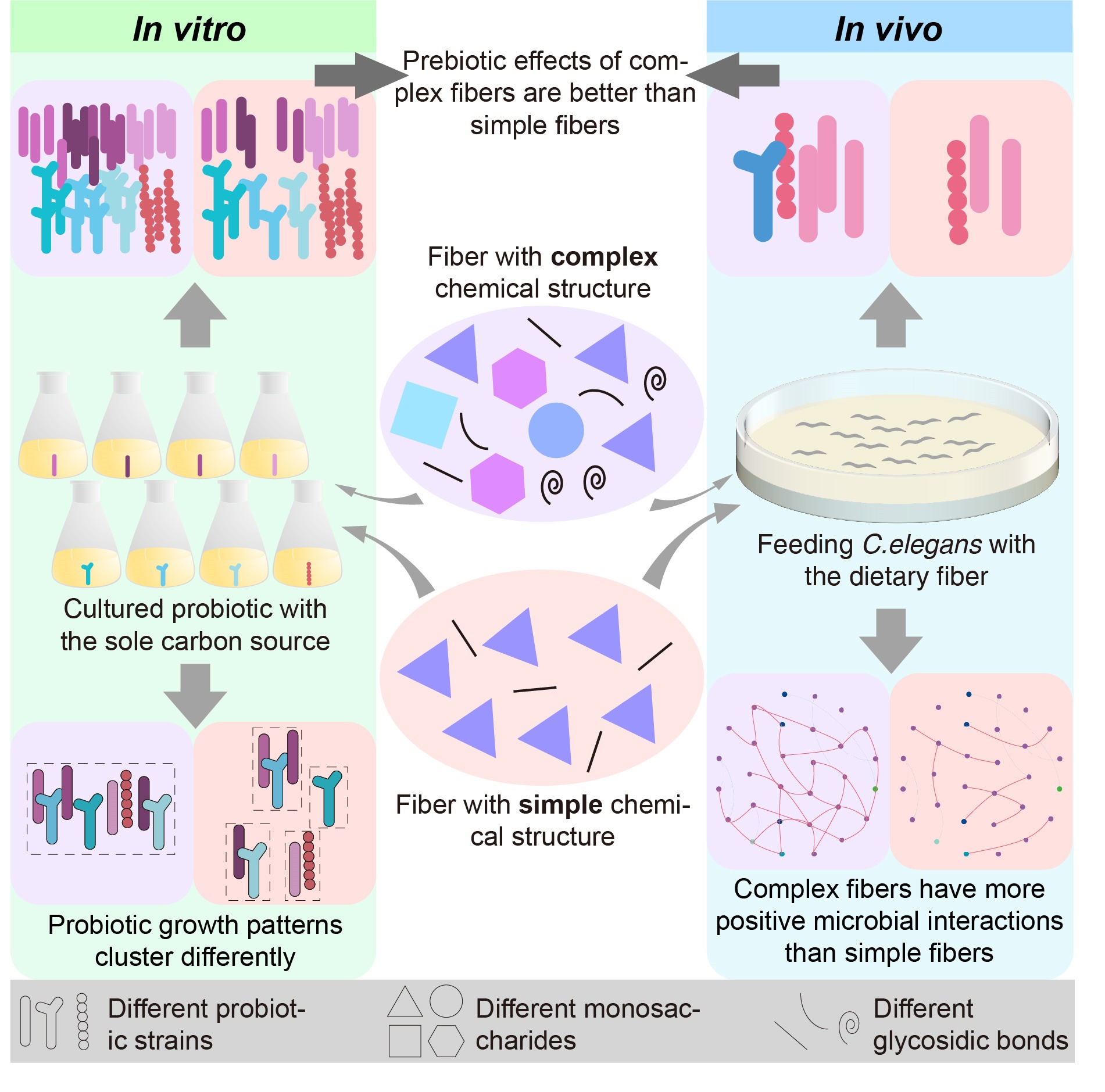
The monosaccharide composition and glycosidic bond configuration determine the CAZyme that could hydrolyze it. Thus, bacteria encoding specific CAZyme utilize different structured dietary fibers. Compounded with bacterial interaction, gut microbiota dynamics is synergistically modulated.
Charting the landscape of the environmental exposome
- 2 September 2022
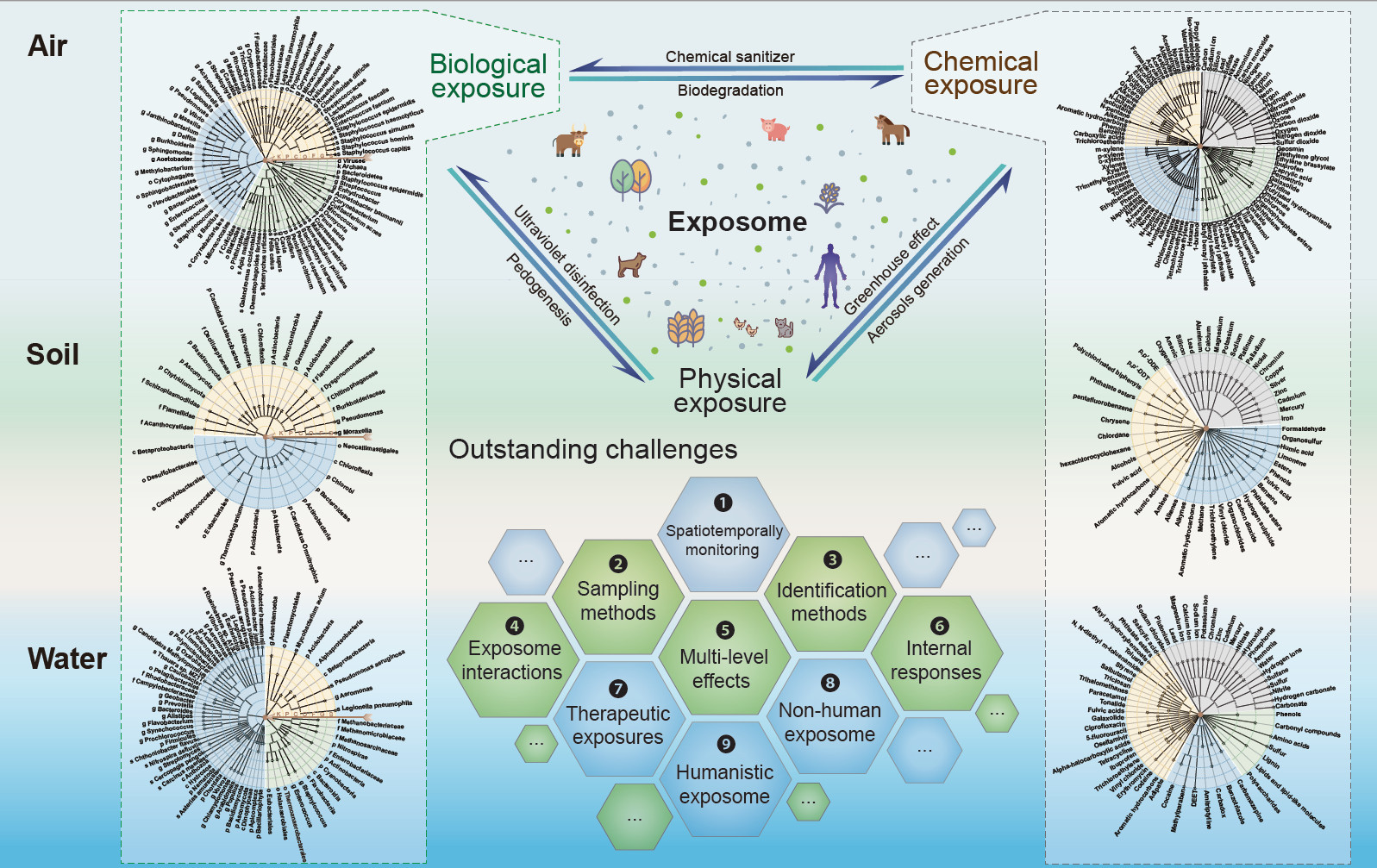
We describe the biological and chemical components of the environmental exposomes in three major environmental matrices that are highly relevant to human and social-economical health—air, soil, and water. We discuss how different exposome components can interact with each other. Finally, we propose a list of outstanding challenges to be tackled to push the field forward.
Gut microbiota-stem cell niche crosstalk: A new territory for maintaining intestinal homeostasis
- 27 September 2022
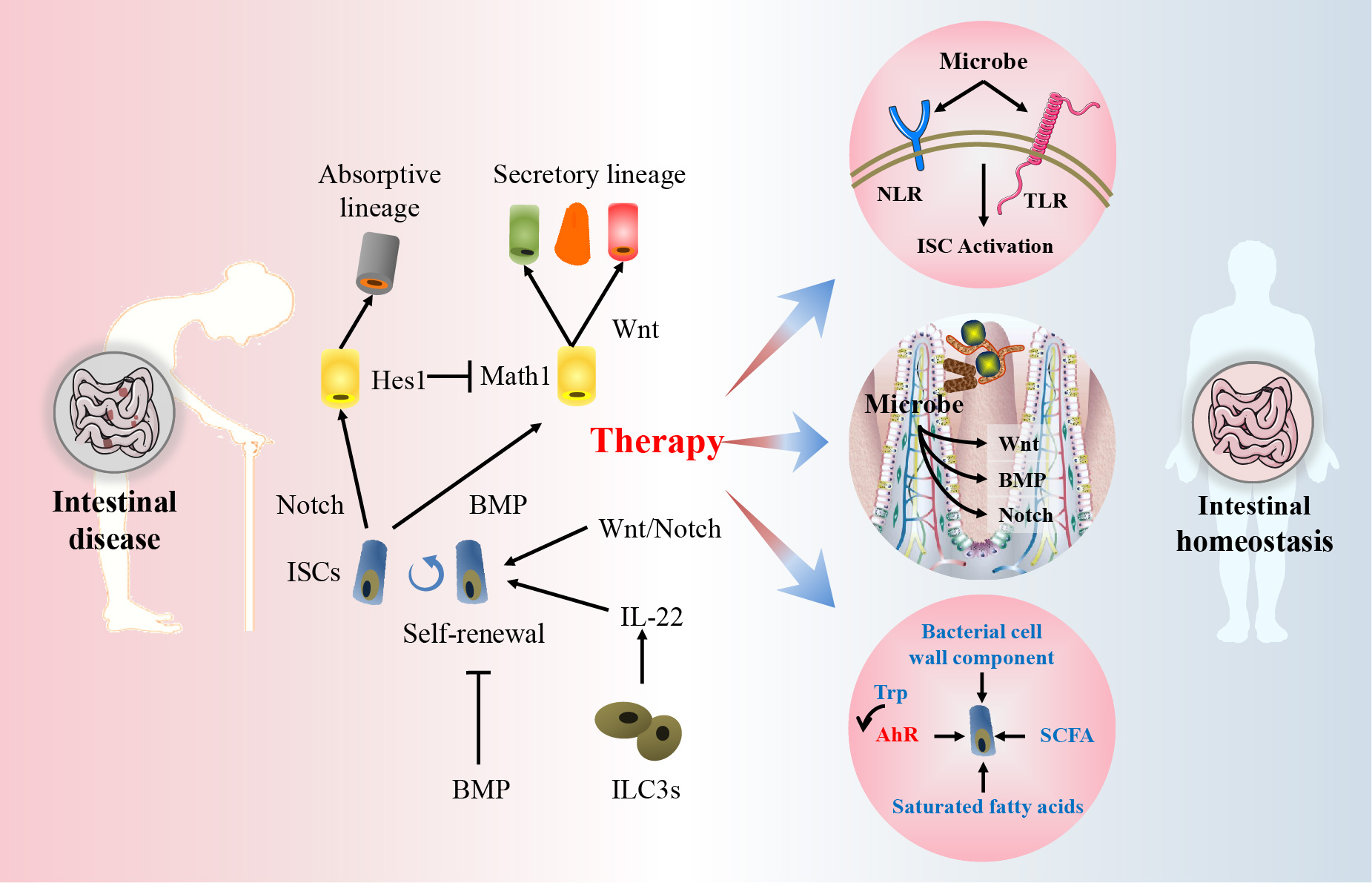
Intestinal stem cells (ISCs) are protected by their niche and are regulated by Wnt, bone morphogenetic protein, and Notch. ISCs and their relationship with intestinal microbiota provide a feasible pathway to alleviate intestinal diseases. Diverse bacteria-related postbiotics regulate ISCs and maintain their homeostasis.
Over two decades of research on the marine RNA virosphere
- 17 October 2022
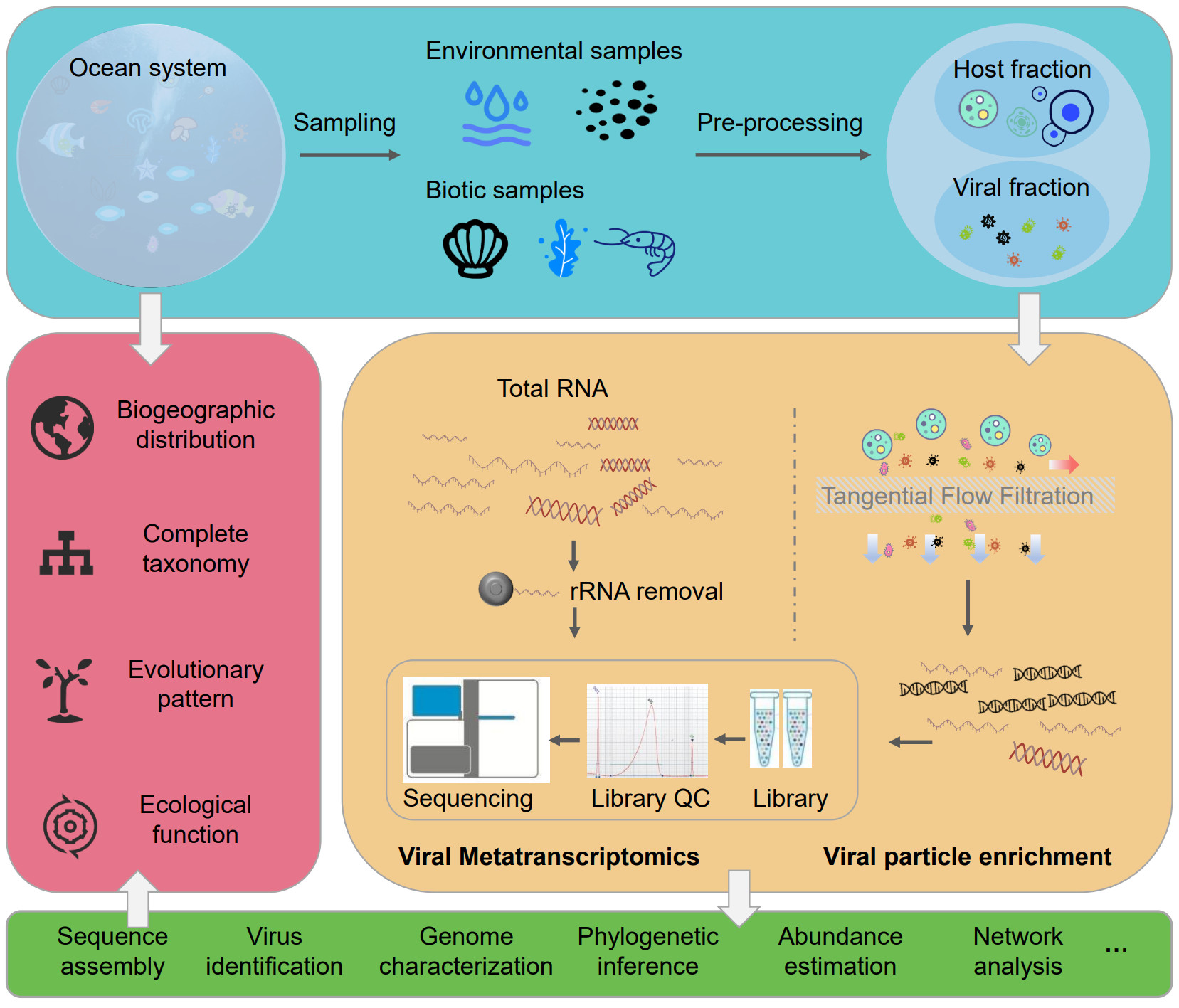
Intestinal stem cells (ISCs) are protected by their niche and are regulated by Wnt, bone morphogenetic protein, and Notch. ISCs and their relationship with intestinal microbiota provide a feasible pathway to alleviate intestinal diseases. Diverse bacteria-related postbiotics regulate ISCs and maintain their homeostasis.
Novel RNA viruses in oysters revealed by virome
- 08 November 2022
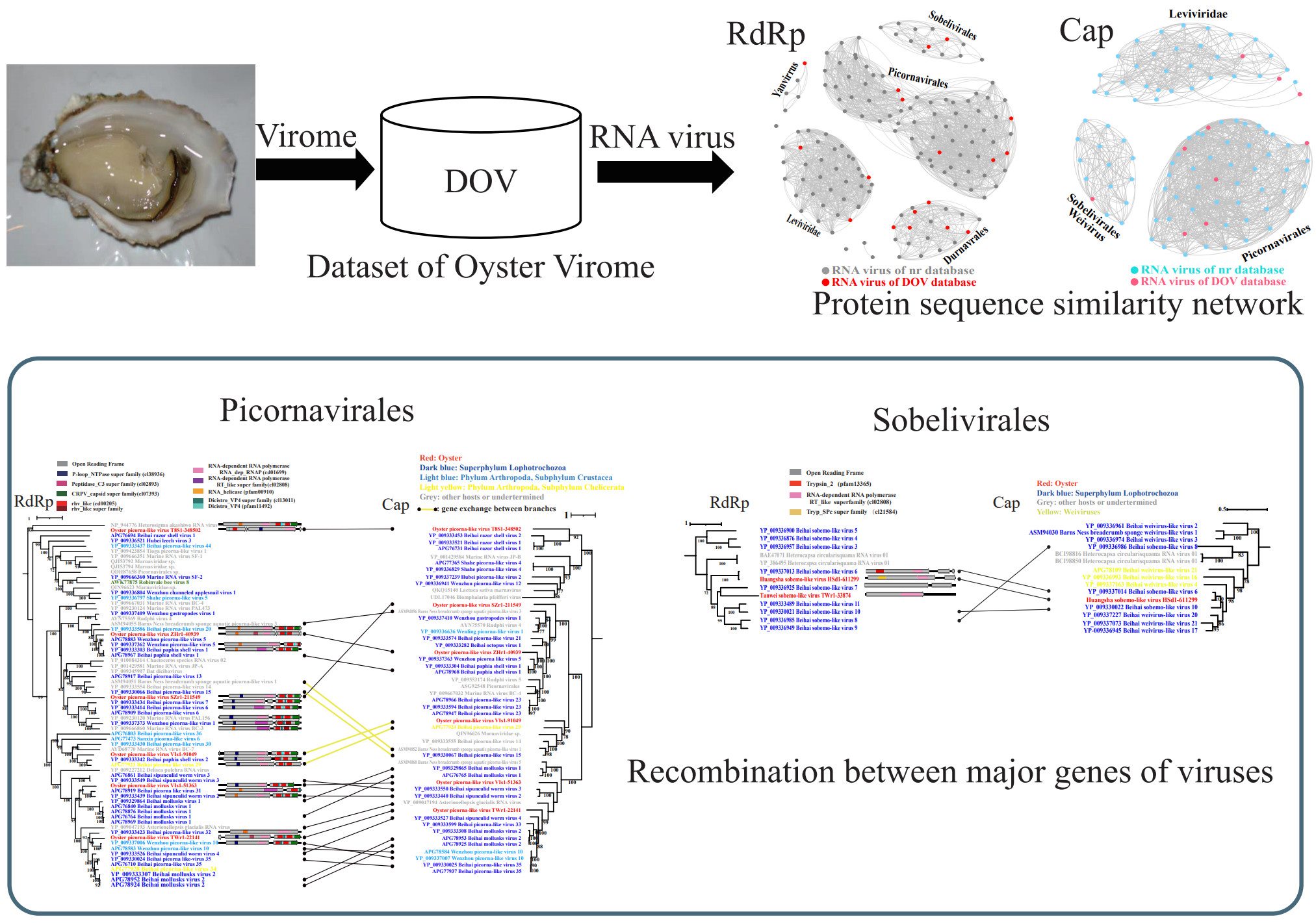
Eighteen novel RNA viruses were found in Crassostrea hongkongensis. Phylogenic analysis shows evidence of recombination between major genes of viruses. Picobirnaviruses are ubiquitous and abundant in oysters.
Donors' experiences and attitudes of fecal microbiota transplantation: An empirical bioethics study from China
- 21 November 2022
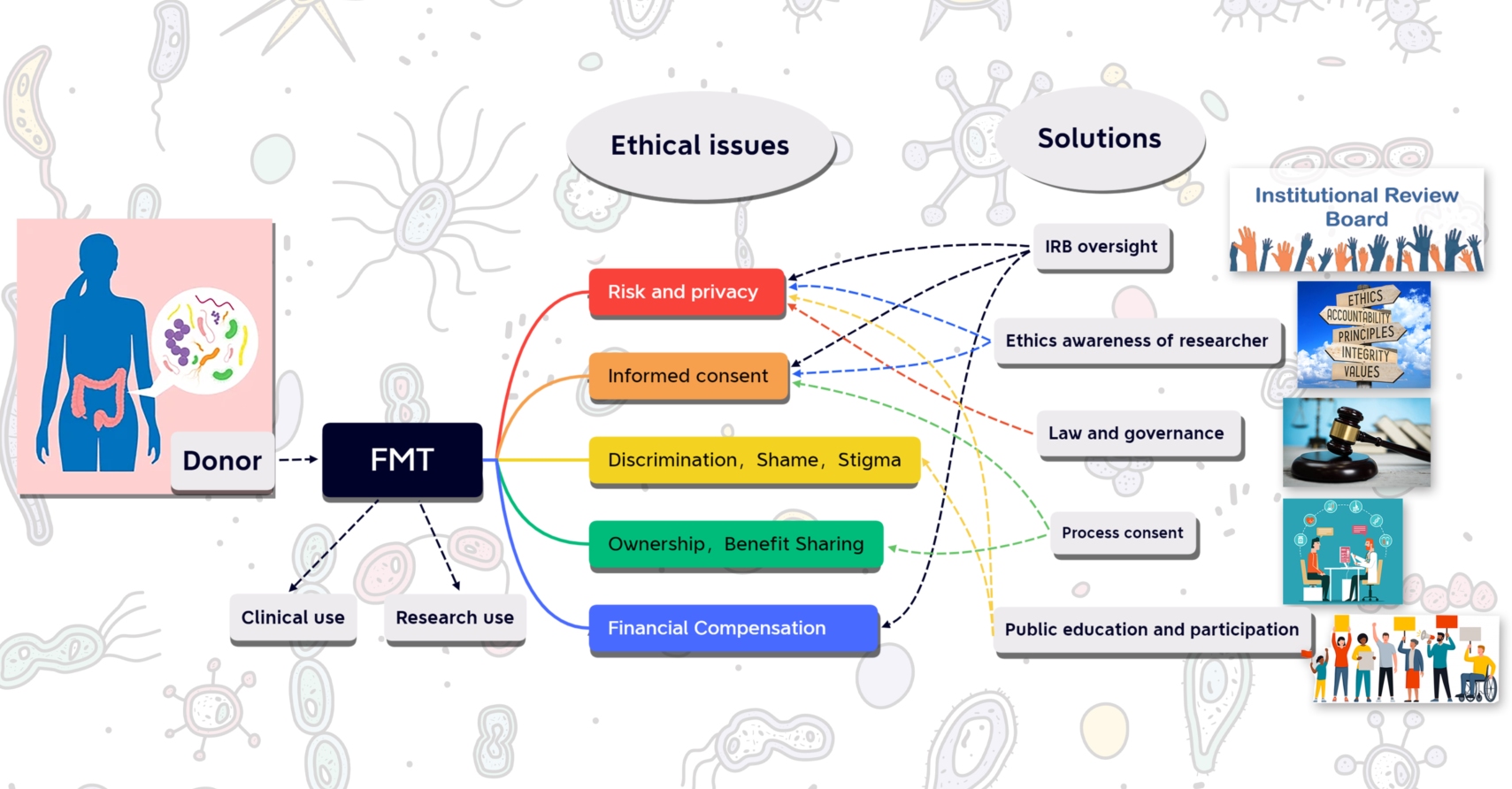
Donor participation is a critical part of ensuring the development of human microbiome research and the clinical application of fecal microbiota transplantation (FMT). Most FMT donors are still not sufficiently aware of the risks associated with the act of donating gut microbiota, especially the risk of data privacy disclosure. Enhanced awareness of the moral responsibility of the researchers and ethical oversight by ethics committees are needed.
Geographic imprint and ecological functions of the abiotic component of periphytic biofilms
- 25 October 2022
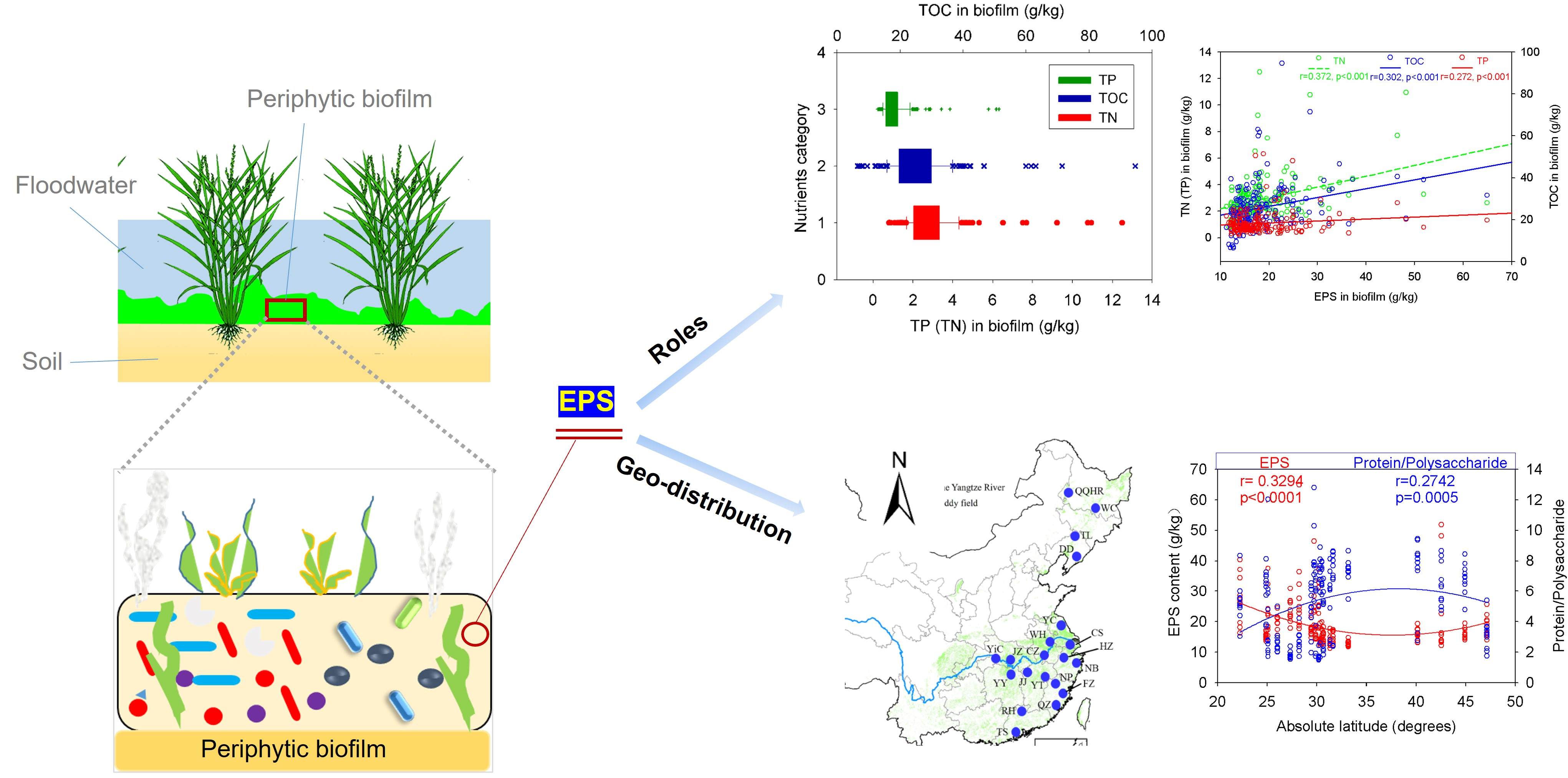
We revealed abiotic components (extracellular polymeric substances, EPSs) in the periphytic biofilms. Further, the effect of the microbial community on the EPS, and the geodistribution patterns and ecological functions of the EPS were studied.
Decoding the role of immune T cells: A new territory for improvement of metabolic-associated fatty liver disease
- 18 January 2023
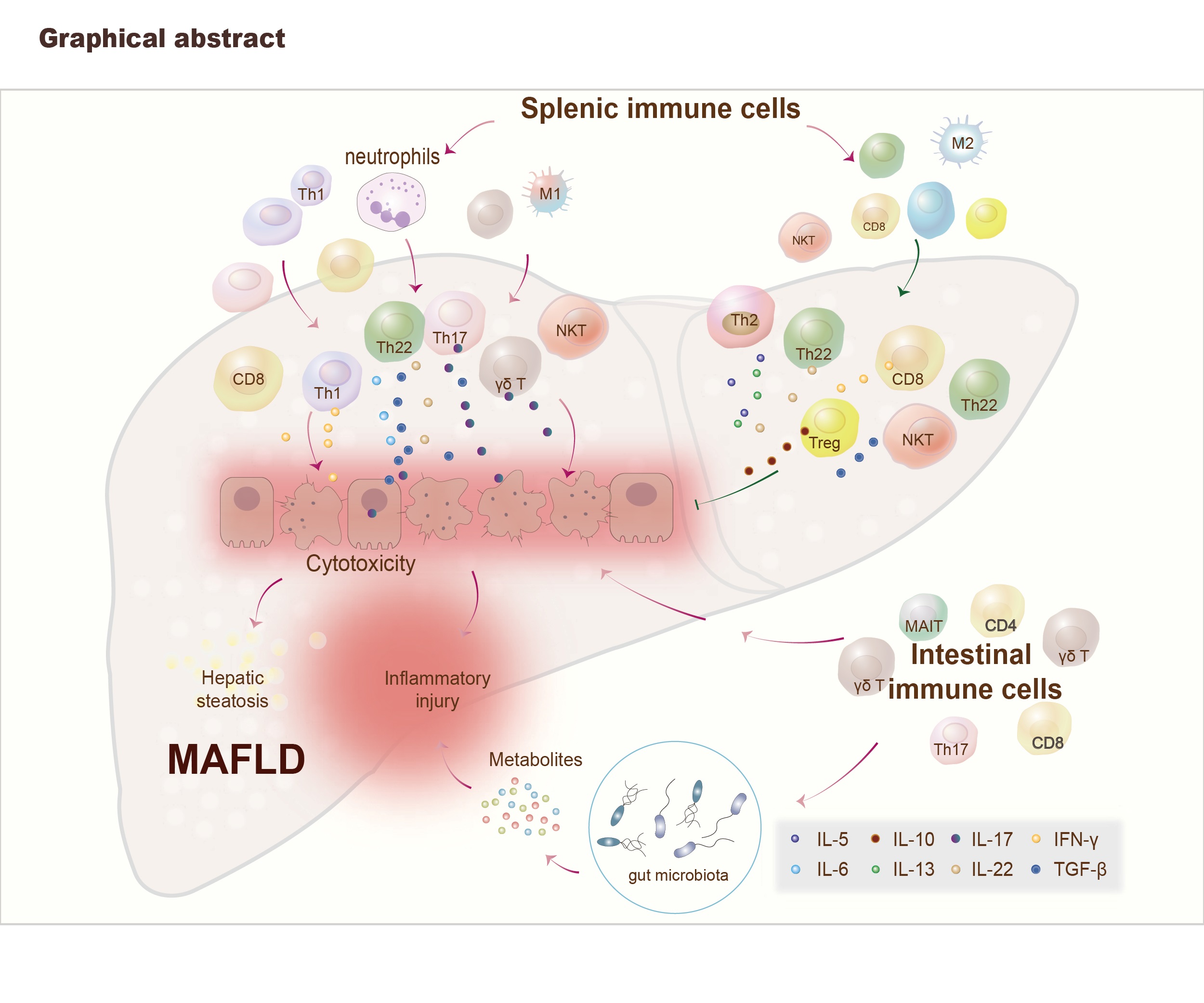
Immune T cells are involved in metabolic-associated fatty liver disease (MAFLD) progression either in a positive or negative manner and the therapeutic methods of MAFLD caused by disordered T cells include gut microbiota regulation, adoptive cell transfer, gene editor microRNAs, drugs (inhibitors/traditional Chinese medicine) therapy.






