
JCVI: A versatile toolkit for comparative genomics analysis
- 12 June 2024
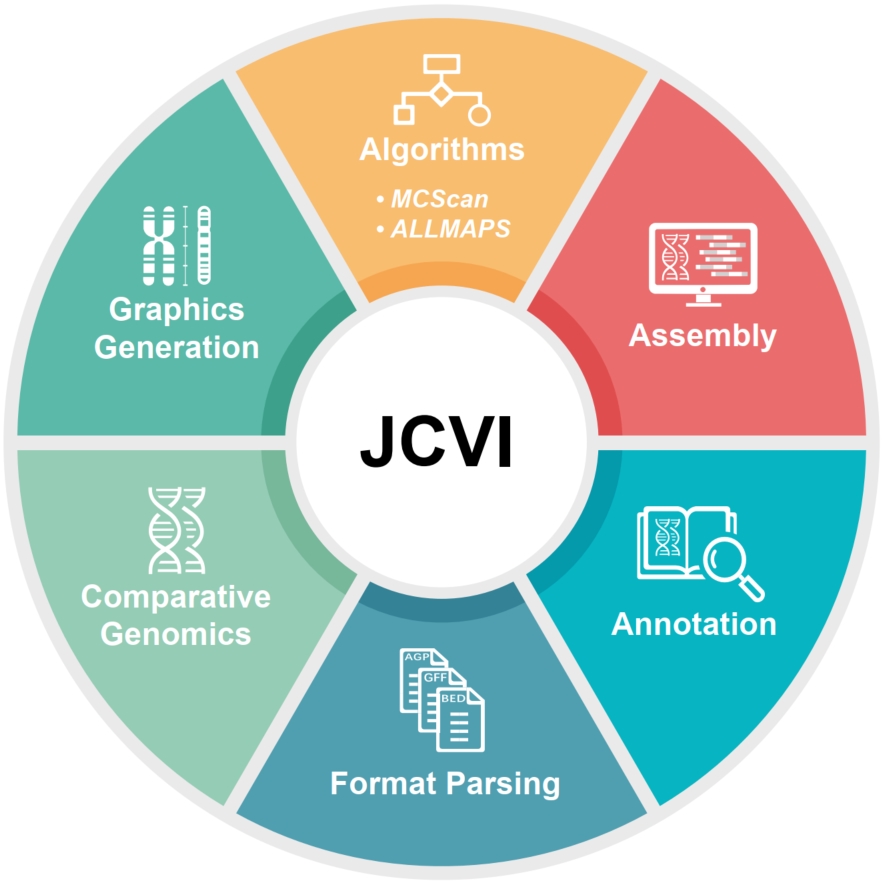
The JCVI library contains a set of computational tools that are often used in tasks covering genome assembly, annotation, and comparative genomics. Engineered with a focus on versatility, the library incorporates modules for algorithms, format parsing, and graphics generation, enabling seamless integration into diverse research workflows.
Genetic manipulations of nonmodel gut microbes
- 23 June 2024
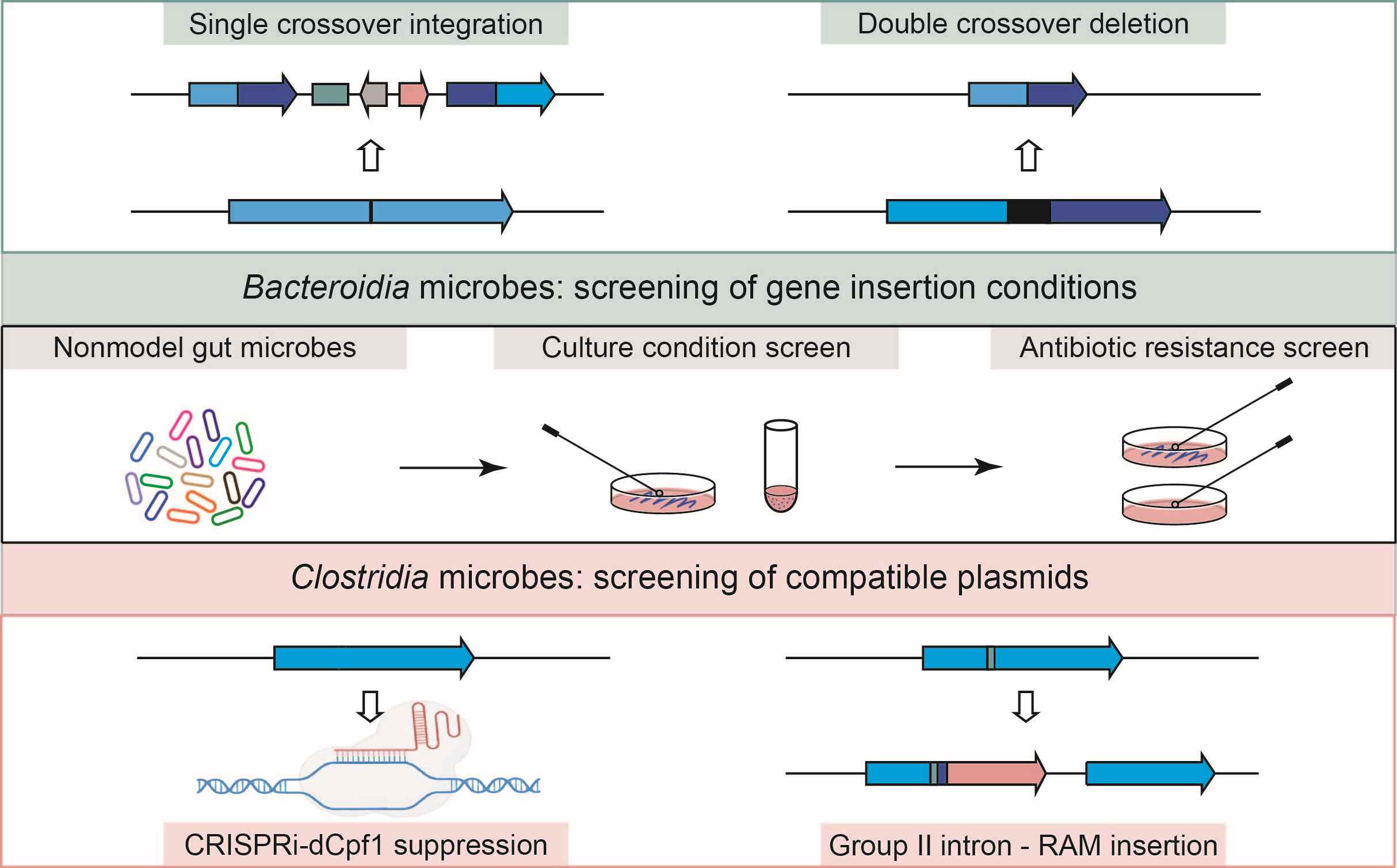
Hundreds of microbiota gene expressions are significantly different between healthy and diseased humans. The “bottleneck” preventing a mechanistic dissection of how they affect host biology/disease is that many genes are encoded by nonmodel gut commensals and not genetically manipulatable. Approaches to efficiently identify their gene transfer methodologies and build their gene manipulation tools would enable mechanistic dissections of their impact on host physiology. This paper will introduce a step-by-step protocol to identify gene transfer conditions and build the gene manipulation tools for nonmodel gut microbes, focusing on Gram-negative Bacteroidia and Gram-positive Clostridia organisms. This protocol enables us to identify gene transfer methods and develop gene manipulation tools without prior knowledge of their genome sequences, by targeting bacterial 16s ribosomal RNAs or expanding their compatible replication origins combined with clustered regularly interspaced short palindromic repeats machinery. Such an efficient and generalizable approach will facilitate functional studies that causally connect gut microbiota genes to host diseases.
Duck gut metagenome reveals the microbiome signatures linked to intestinal regional, temporal development, and rearing condition
- 14 May 2024
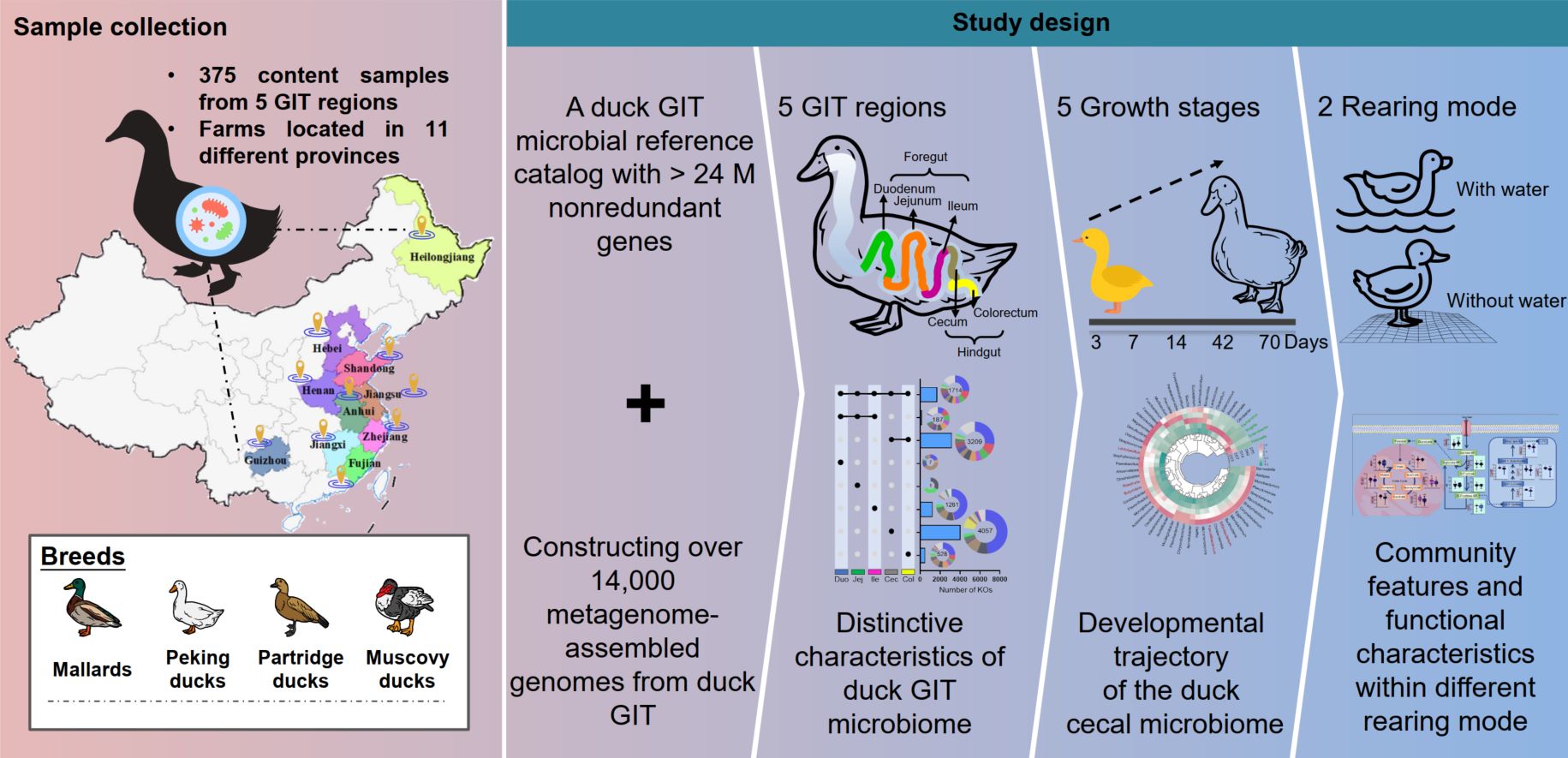
In this study, we constructed the first relatively comprehensive duck gut microbial gene catalog and metagenome-assembled genomes using 375 duck gastrointestinal tract metagenomic samples from four different duck breeds across five intestinal segments under two distinct rearing conditions. Furthermore, the present study expands the understanding of the duck microbiome signatures linked to intestinal regional, temporal development, and rearing conditions in ducks, highlighting the significant impact of microbiota on poultry health and production.
Deep learning enhancing guide RNA design for CRISPR/Cas12a-based diagnostics
- 15 June 2024
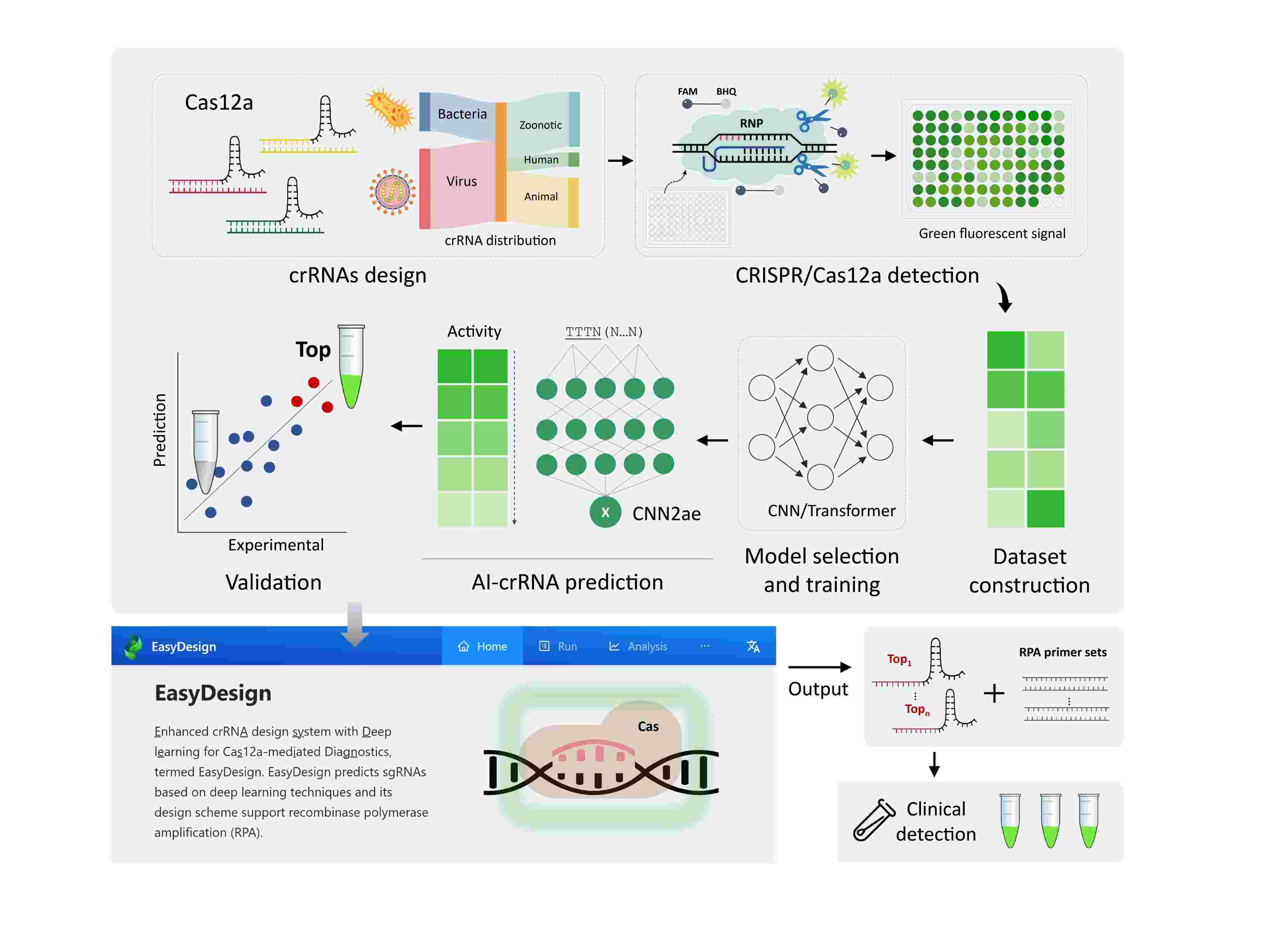
Advanced crRNA Design System: Developed EasyDesign, utilizing a convolutional neural network (CNN) trained with over 11,000 diagnostic-target pairs to enable the creation of highly sensitive crRNAs for Cas12a-based nucleic acid diagnostics. Proven Predictive Capabilities: EasyDesign demonstrates superior predictive performance for crRNA-mediated Cas12a detection, validated through its successful application in designing diagnostics for a variety of viruses in clinical settings. User-friendly web platform: Features an intuitive web platform that streamlines the creation of CRISPR/Cas12a-based diagnostic tools, thereby enhancing accessibility and usability for both researchers and clinicians.
Prophylactic supplementation with Bifidobacterium infantis or its metabolite inosine attenuates cardiac ischemia/reperfusion injury
- 02 July 2024
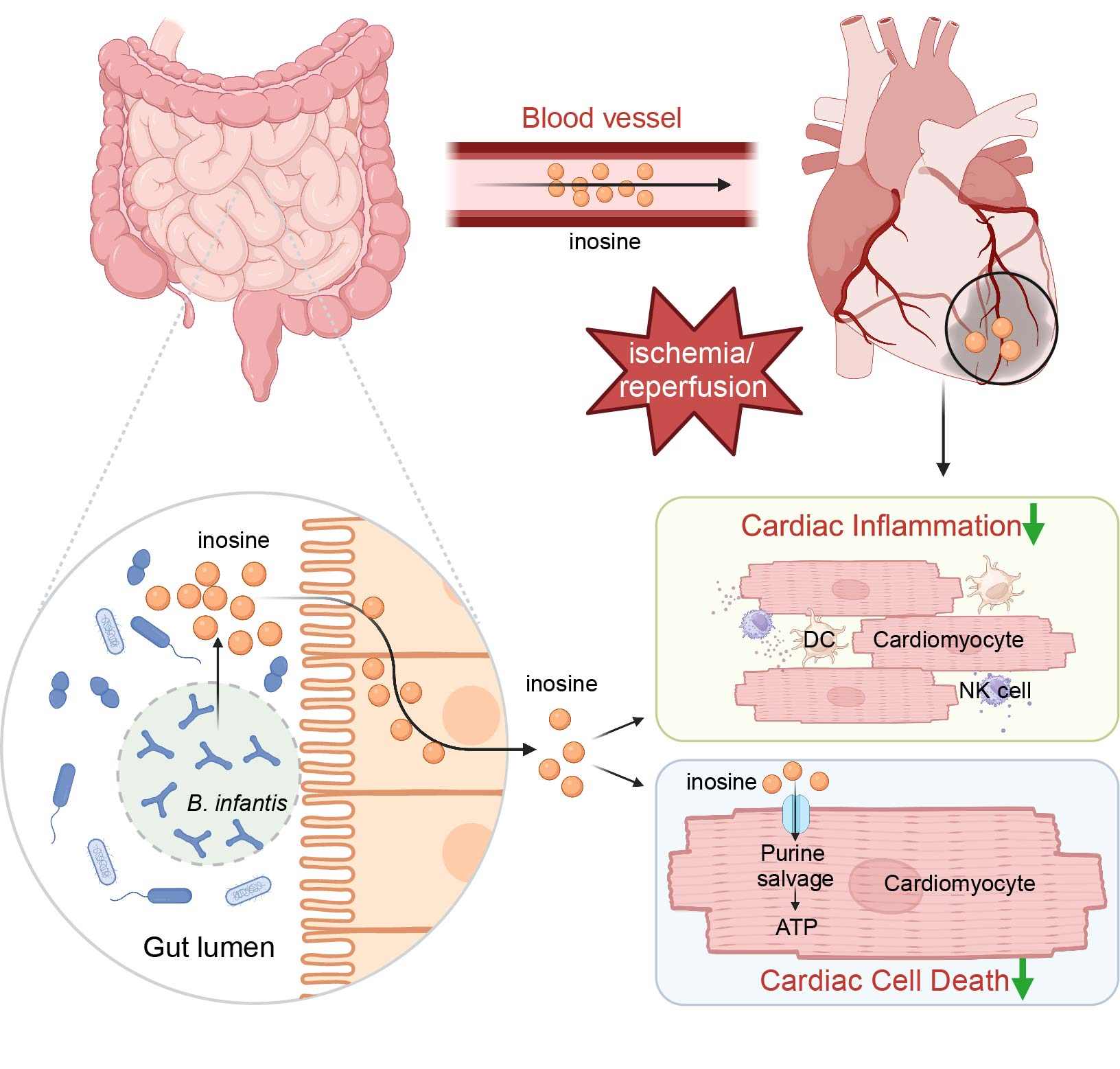
In this study, we found that the prophylactic administration of a well-known probiotic, Bifidobacterium infantis (B. infantis), exhibited cardioprotective effects against myocardial ischemia–reperfusion (I/R) injury, which was recapitulated by its metabolite inosine. Specifically, inosine suppressed cardiac inflammation by reducing the production of pro-inflammatory cytokines and decreasing the numbers of dendritic cells and natural killer cells after I/R. Additionally, inosine attenuated cell death by serving as an alternative carbon source for adenosine triphosphate generation through the purine salvage pathway in stressed myocytes and in I/R-injured mouse hearts. Collectively, our findings indicate that B. infantis or its metabolite inosine exerts pleiotropic cardioprotective effects against I/R, suggesting prophylactic therapeutic options for acute ischemic cardiac injury.
Cell-type-specific expression analysis of liver transcriptomics with clinical parameters to decipher the cause of intrahepatic inflammation in chronic hepatitis B
- 04 July 2024
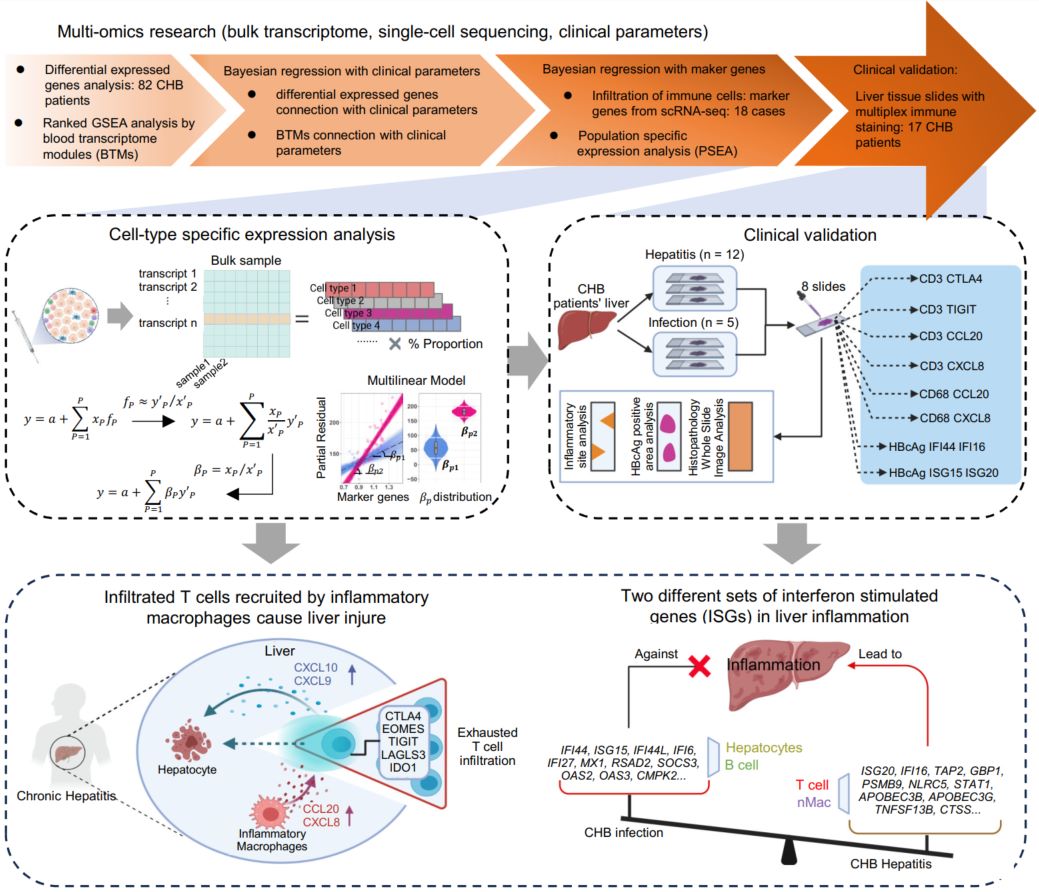
This study integrates liver bulk transcriptomic data, single-cell sequencing data, and clinical data to analyze the factors that induce hepatic inflammation in chronic hepatitis B from a multi-omics perspective by Bayesian regression. Macrophages secrete chemokines like CCL20 and CXCL8 to recruit immune-exhausted T lymphocytes (CTLA4, TIGIT) into liver tissue. Innate immunity within hepatocytes is suppressed, impeding interferon-stimulated genes from initiating antiviral effects. Activation of innate immune pathways in infiltrating T cells and macrophages further exacerbates inflammation formation.
Intestinal microbiota by angiotensin receptor blocker therapy exerts protective effects against hypertensive damages
- 18 July 2024
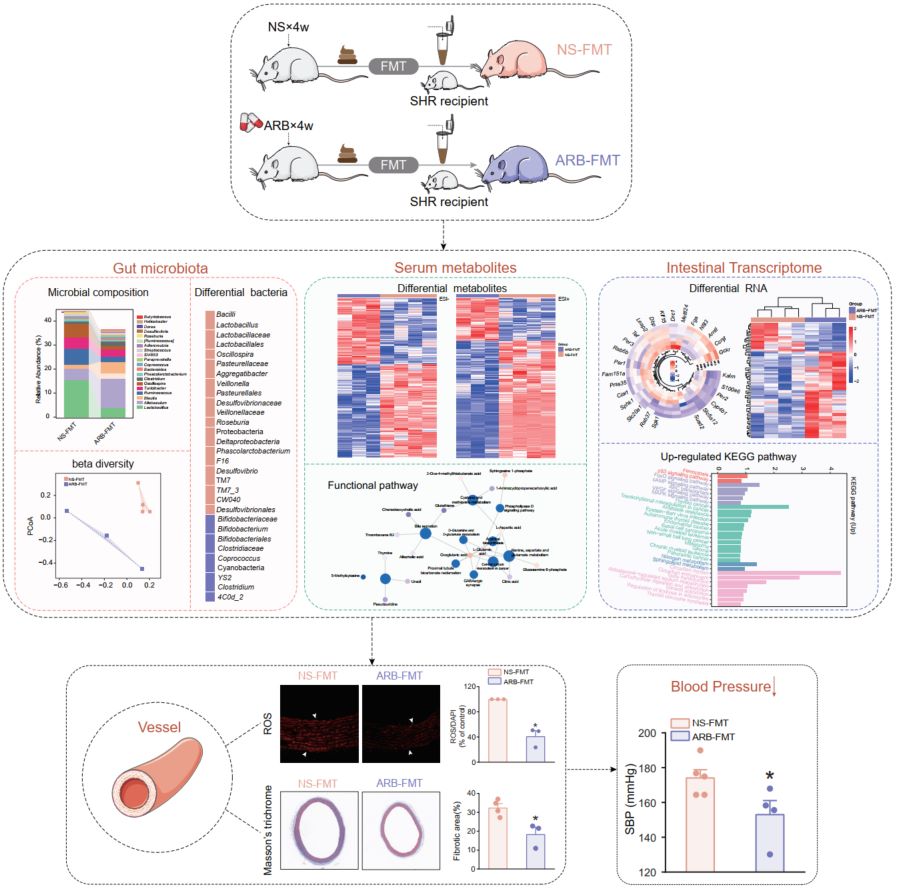
16S rRNA sequencing, liquid chromatography-tandem mass spectrometry, RNA sequencing and immunofluorescence staining were combined to assess the role of ARB-modified gut microbes. ARB-modified gut microbiota exert protective roles in hypertension, vascular remodeling and injury, metabolic abnormality and intestinal dysfunctions, suggesting a pivotal role in mitigating hypertension and providing insights into the cross-talk between antihypertensive medicines and the gut microbiome.
Deciphering functional groups of rumen microbiome and their underlying potentially causal relationships in shaping host traits
- 15 July 2024
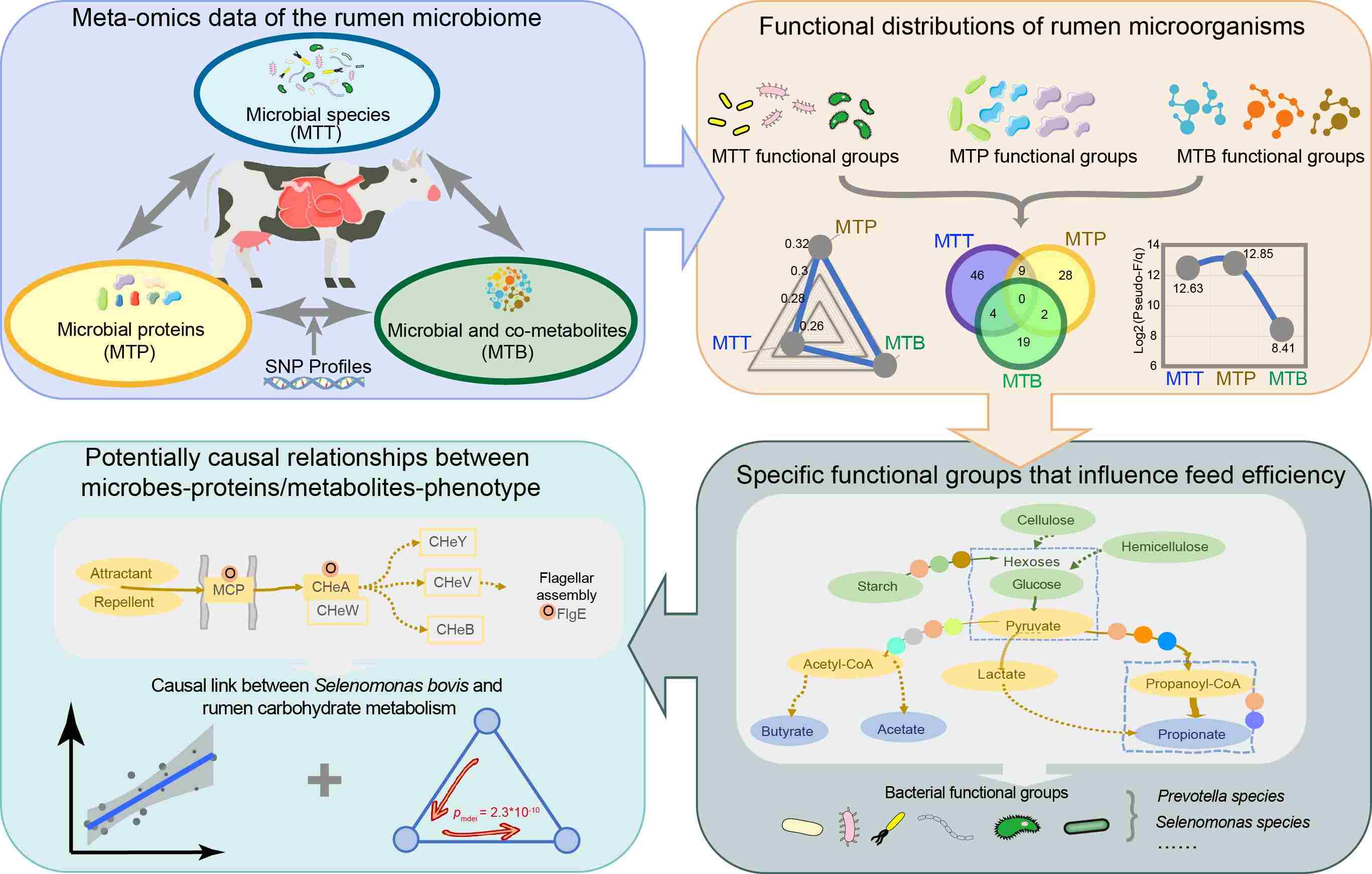
In this study, we focused on the rumen metaproteome, combining it with metatranscriptome and metabolome data to identify the active functional distributions of rumen microorganisms and specific functional groups that influence feed efficiency. By integrating host genetics data, we established potentially causal relationships between microbes, proteins/metabolites, and phenotypes. We identified specific patterns in which functional groups of rumen microorganisms influence host feed efficiency. We found a causal link between Selenomonas bovis and rumen carbohydrate metabolism, potentially mediated by bacterial chemotaxis and a two-component regulatory system, impacting feed utilization efficiency in dairy cows. Our study on nutrient utilization functional groups in the rumen of high-feed-efficiency dairy cows, along with identifying key microbiota functional proteins and their potential causal relationships, will help move from correlation to causation in rumen microbiome research. This will enable precise regulation of the rumen microbiota for optimized ruminant production.
Single-cell landscape revealed immune characteristics associated with disease phases in brucellosis patients
- 15 July 2024
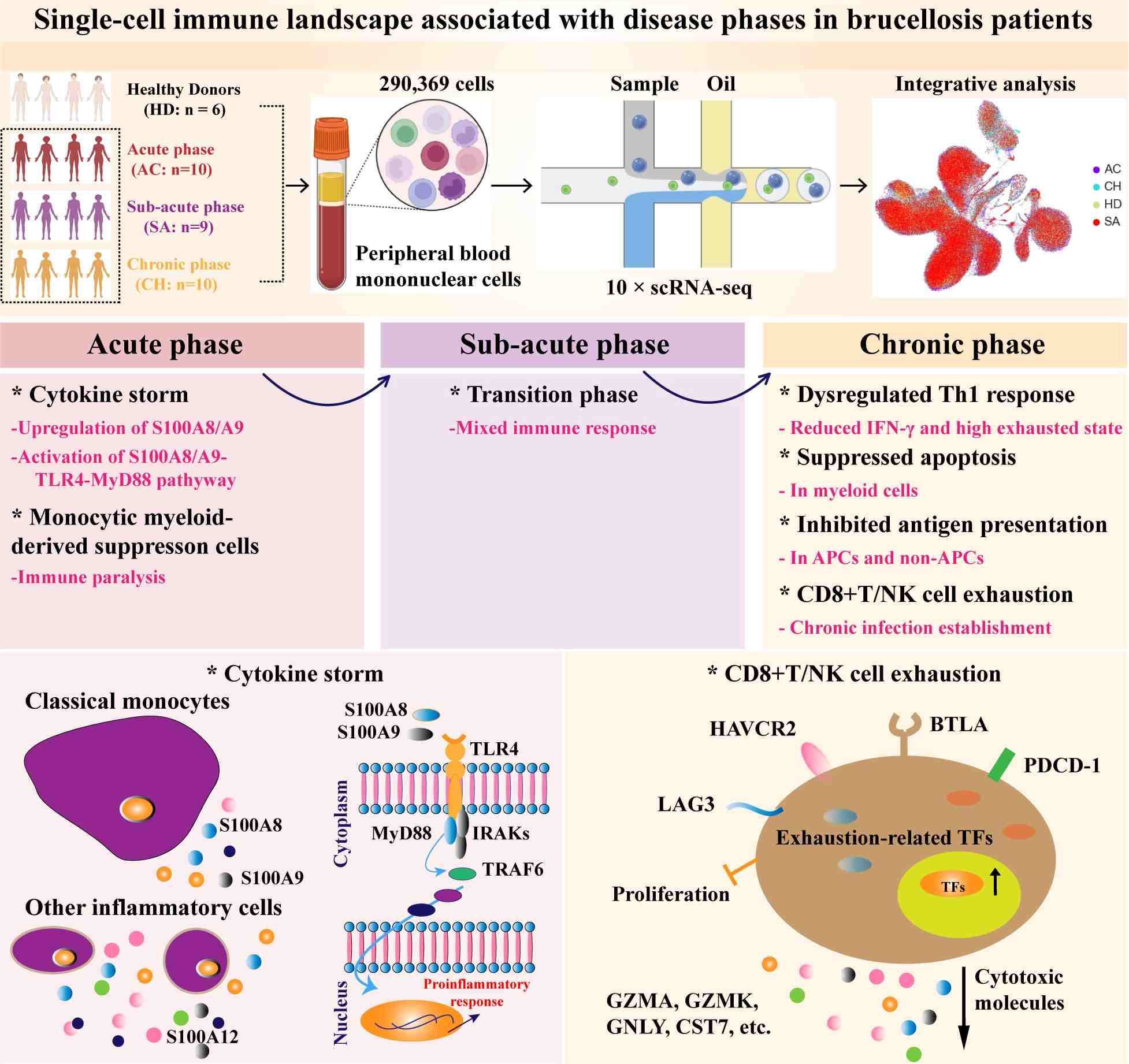
Single-cell RNA sequencing of 290,369 cells from 29 brucellosis patients and 6 healthy donors reveals distinct immune profiles across acute, subacute, and chronic phases of the disease. Acute patients experience a cytokine storm triggered by classical monocytes, potentially mediated by the S100A8/A9-TLR4-MyD88 signaling pathway. Chronic patients exhibit a dysregulated Th1 response, characterized by reduced expression of IFN-γ and Th1-associated gene signatures, alongside T and NK cell exhaustion. This investigation sheds light on the complex immunopathogenesis of brucellosis, providing valuable insights that may facilitate the development of novel therapeutic strategies, particularly for managing chronic infections.
MASS cohort: Multicenter, longitudinal, and prospective study of the role of microbiome in severe pneumonia and host susceptibility
- 25 June 2024
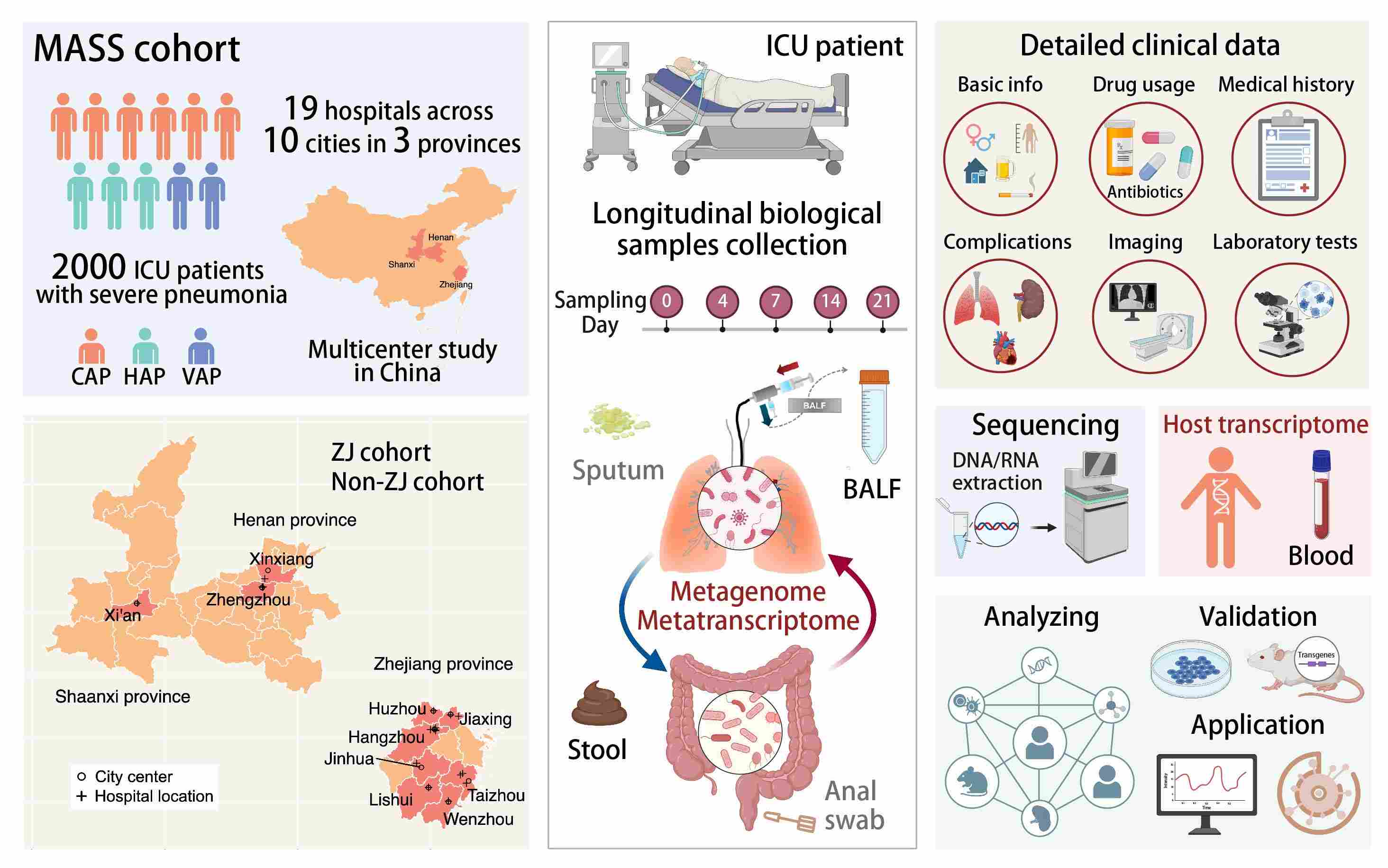
The MASS cohort comprises 2000 ICU patients with severe pneumonia, covering community-acquired pneumonia, hospital-acquired pneumonia, and ventilator-associated pneumonia, sourced from 19 hospitals across 10 cities in three provinces. A wide array of samples including bronchoalveolar lavage fluid, sputum, feces, and whole blood are longitudinally collected throughout patients' ICU stays. The cohort study seeks to uncover the dynamics of lung and gut microbiomes and their associations with severe pneumonia and host susceptibility, integrating deep metagenomics and transcriptomics with detailed clinical data.
BioLadder: A bioinformatic platform primarily focused on proteomic data analysis
- 20 June 2024
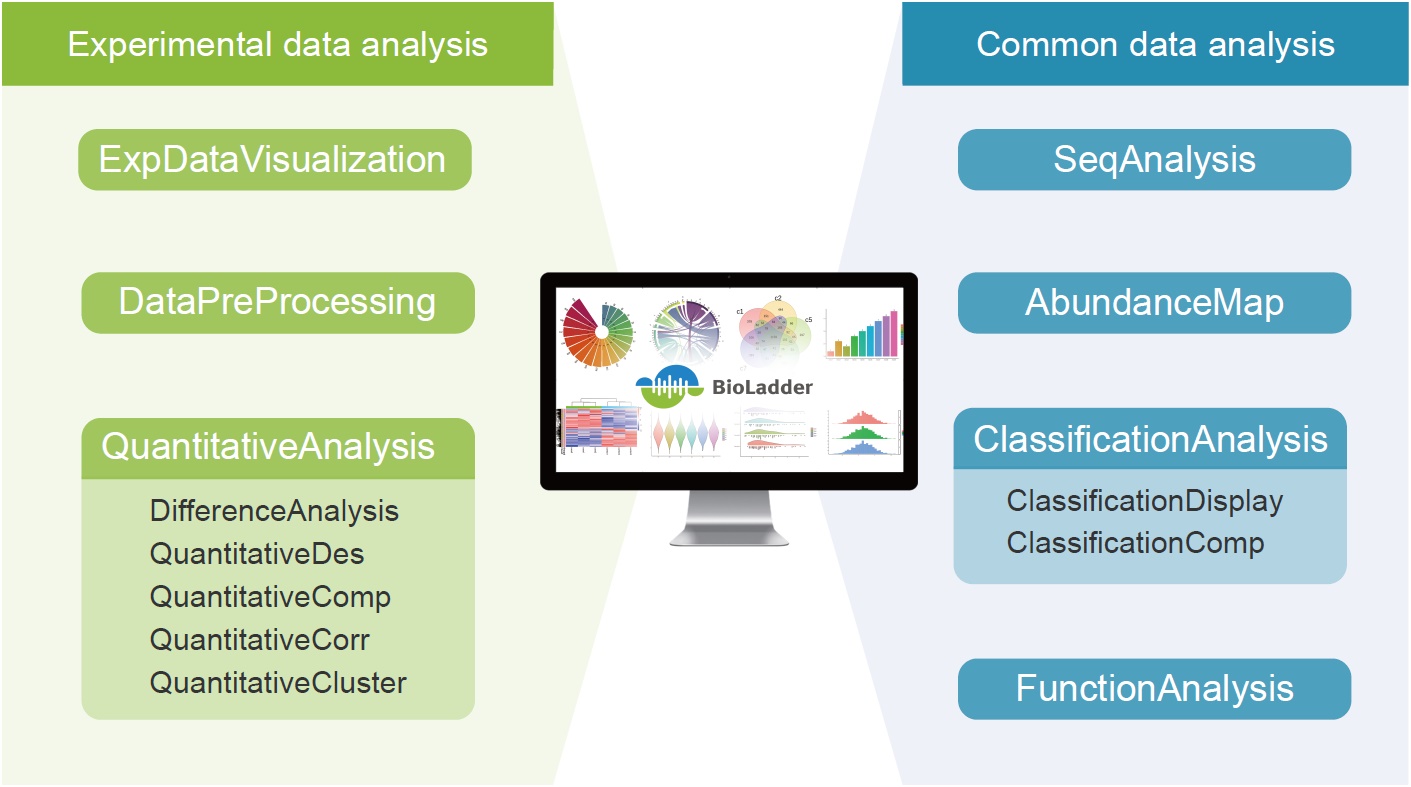
BioLadder (https://www.bioladder.cn/) is an online data analysis platform designed for proteomics research, which includes three classes of experimental data analysis modules and four classes of common data analysis modules. It allows for a variety of proteomics analyses to be conducted easily and efficiently. Additionally, most modules can also be utilized for the analysis of other omics data. To facilitate user experience, we have carefully designed four different kinds of functions for customers to quickly and accurately utilize the relevant analysis modules.
The challenge and opportunity of gut microbiota-targeted nanomedicine for colorectal cancer therapy
- 10 June 2024
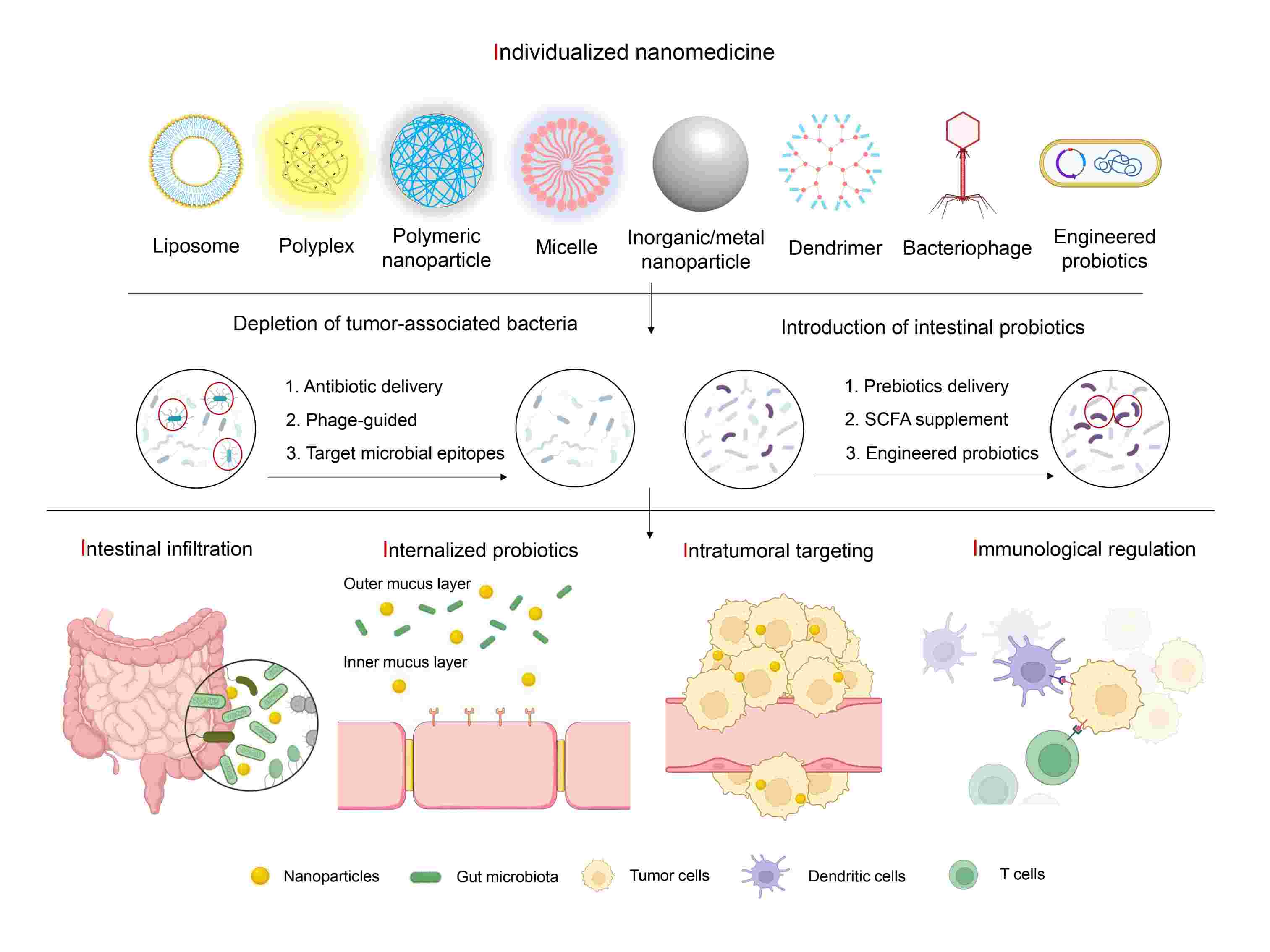
The gut microbiota is an integral component of the colorectal cancer (CRC) microenvironment and is intimately associated with CRC initiation, progression, and therapeutic outcomes. We reviewed recent advancements in utilizing nanotechnology for modulating gut microbiota, discussing strategies and the mechanisms underlying their design. For future nanomedicine design, we propose a 5I principle for individualized nanomedicine in CRC management.
Majorbio Cloud 2024: Update single-cell and multiomics workflows
- 25 June 2024
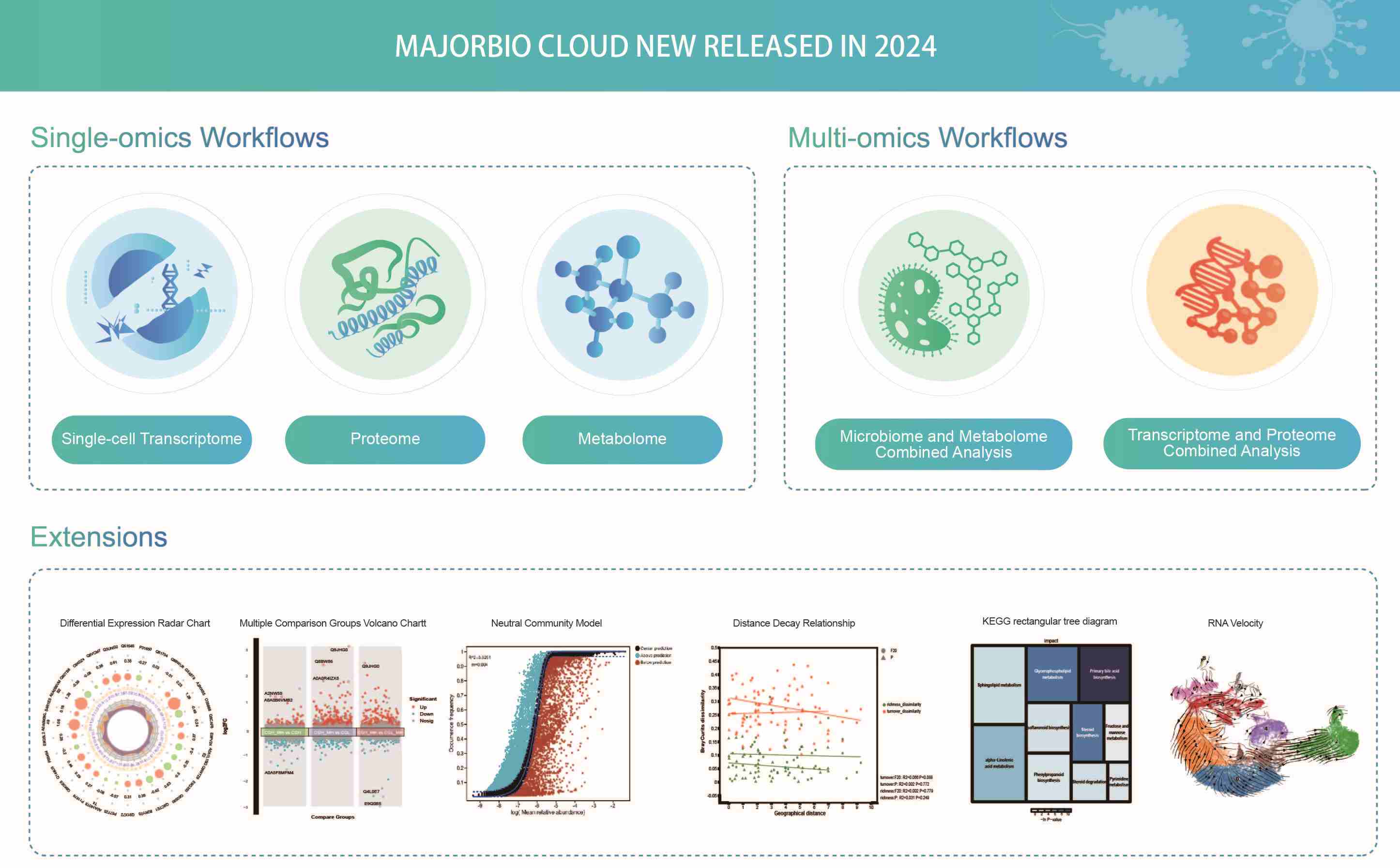
Majorbio Cloud (https://cloud.majorbio.com/) is a one-stop online analytic platform aiming at promoting the development of bioinformatics services, narrowing the gap between wet and dry experiments, and accelerating the discoveries for the life sciences community. In 2024, three single-omics workflows, two multiomics workflows, and extensions were newly released to facilitate omics data mining and interpretation.
Ms gene and Mr gene: Microbial-mediated spatiotemporal communication between plants
- 07 June 2024
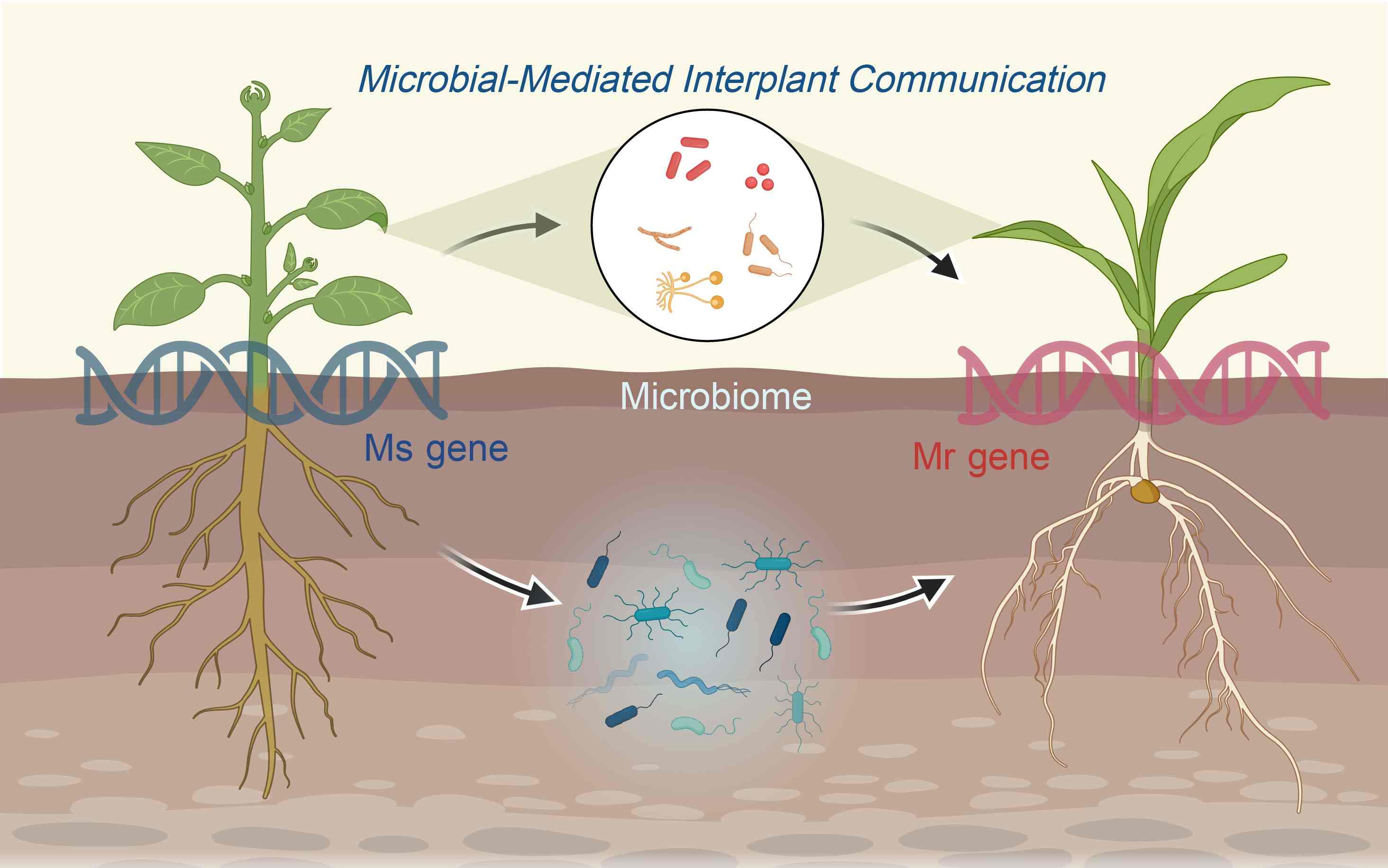
Within dynamic agroecosystems, microbes can act as key intermediaries, facilitating spatiotemporal communication among plants. Future research could categorize key plant genes involved in plant–microbe interactions into microbiome-shaping genes (Ms genes) and microbiome-responsive genes (Mr genes), potentially leading to the construction of spatiotemporal molecular networks with microbes as intermediaries.
Immune-enriched phyllosphere microbiome in rice panicle exhibits protective effects against rice blast and rice false smut diseases
- 15 July 2024
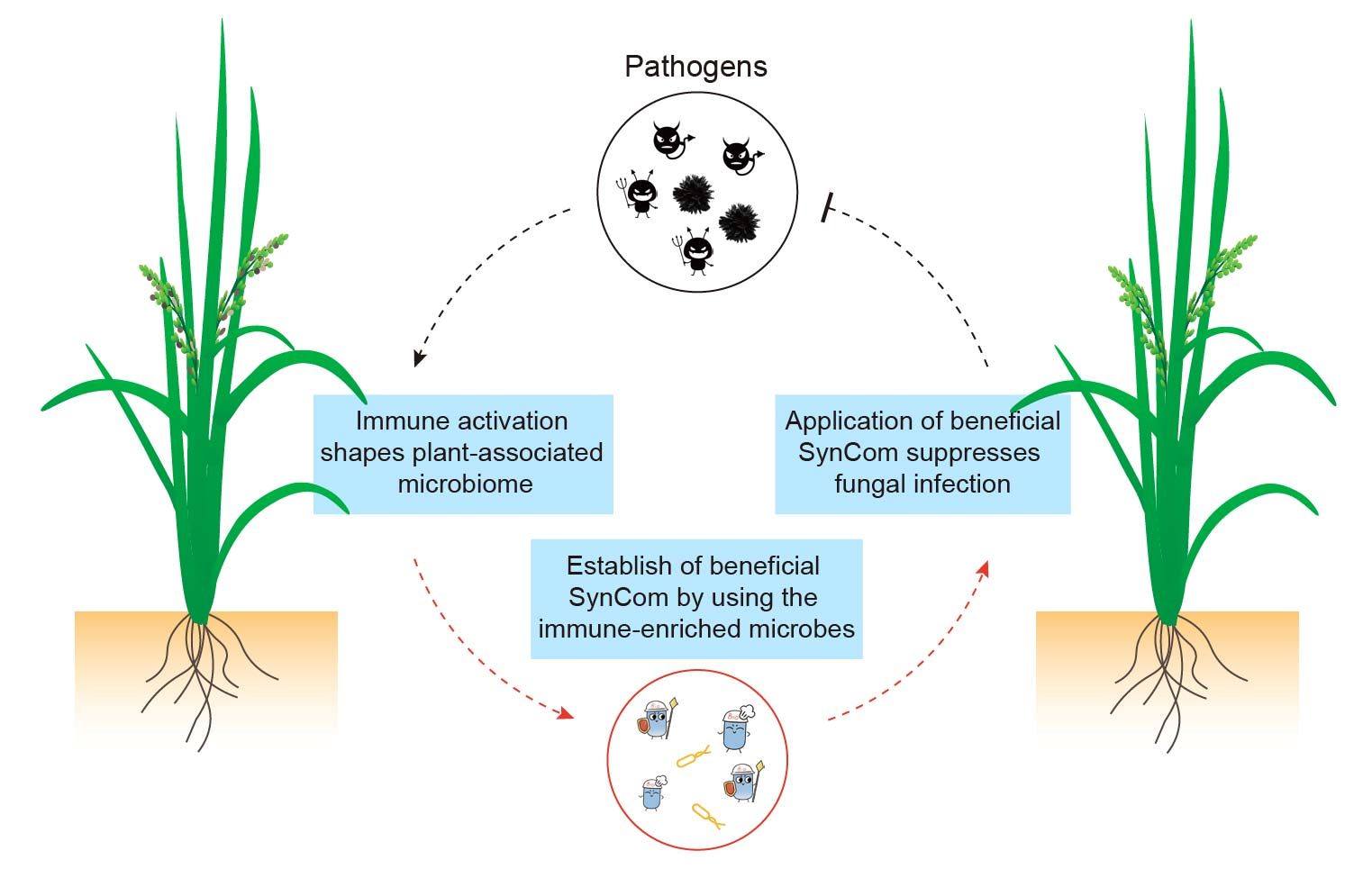
Activation of immune responses leads to an enrichment of beneficial microbes in rice panicle. We therefore selected the enriched operational taxonomy units (OTUs) exhibiting direct suppression effects on fungal pathogens, and established a simplified synthetic community (SynCom) which consists of three beneficial microbes. Application of this SynCom exhibits protective effect against fungal pathogen infection in rice.
Smaller microorganisms outcompete larger ones in resistance and functional effects under disturbed agricultural ecosystems
- 23 June 2024
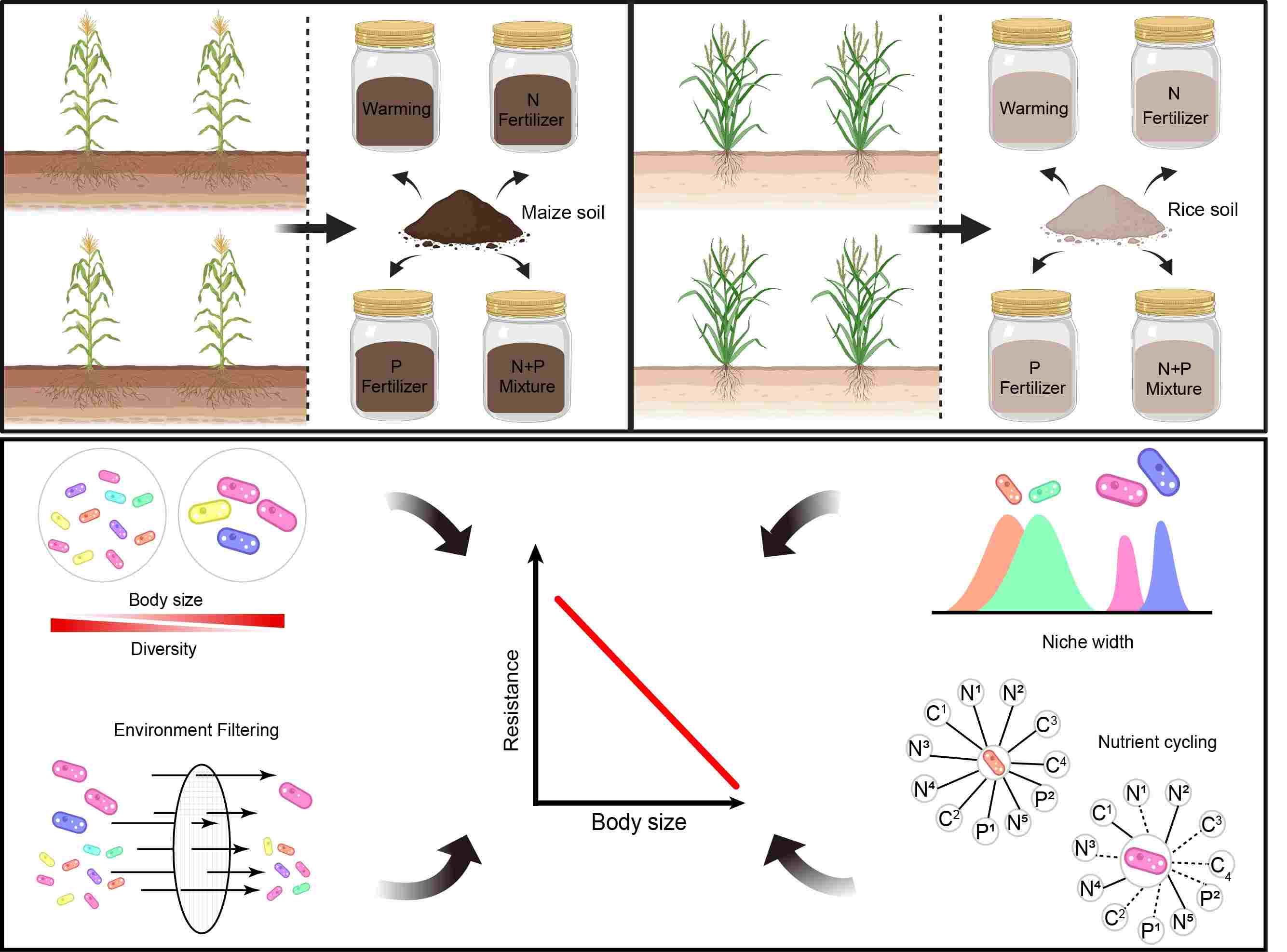
Body size is a key ecological trait of soil microorganisms related to their adaptation to environmental changes. In this study, we reveal that the smaller microorganisms show stronger community resistance than larger organisms in both maize and rice soil. Compared with larger organisms, smaller microorganisms have higher diversity and broader niche breadth to deploy survival strategies, because of which they are less affected by environmental selection and thus survive in complex and various kinds of environments. In addition, the strong correlation between smaller microorganisms and ecosystem functions reflects their greater metabolic flexibility and illustrates their significant roles in adaptation to continuously changing environments. This research highlights the importance of body size in maintaining stability of the soil microbiome and forecasting agroecosystem dynamics under environmental disturbances.
A universal oral microbiome-based signature for periodontitis
- 12 June 2024
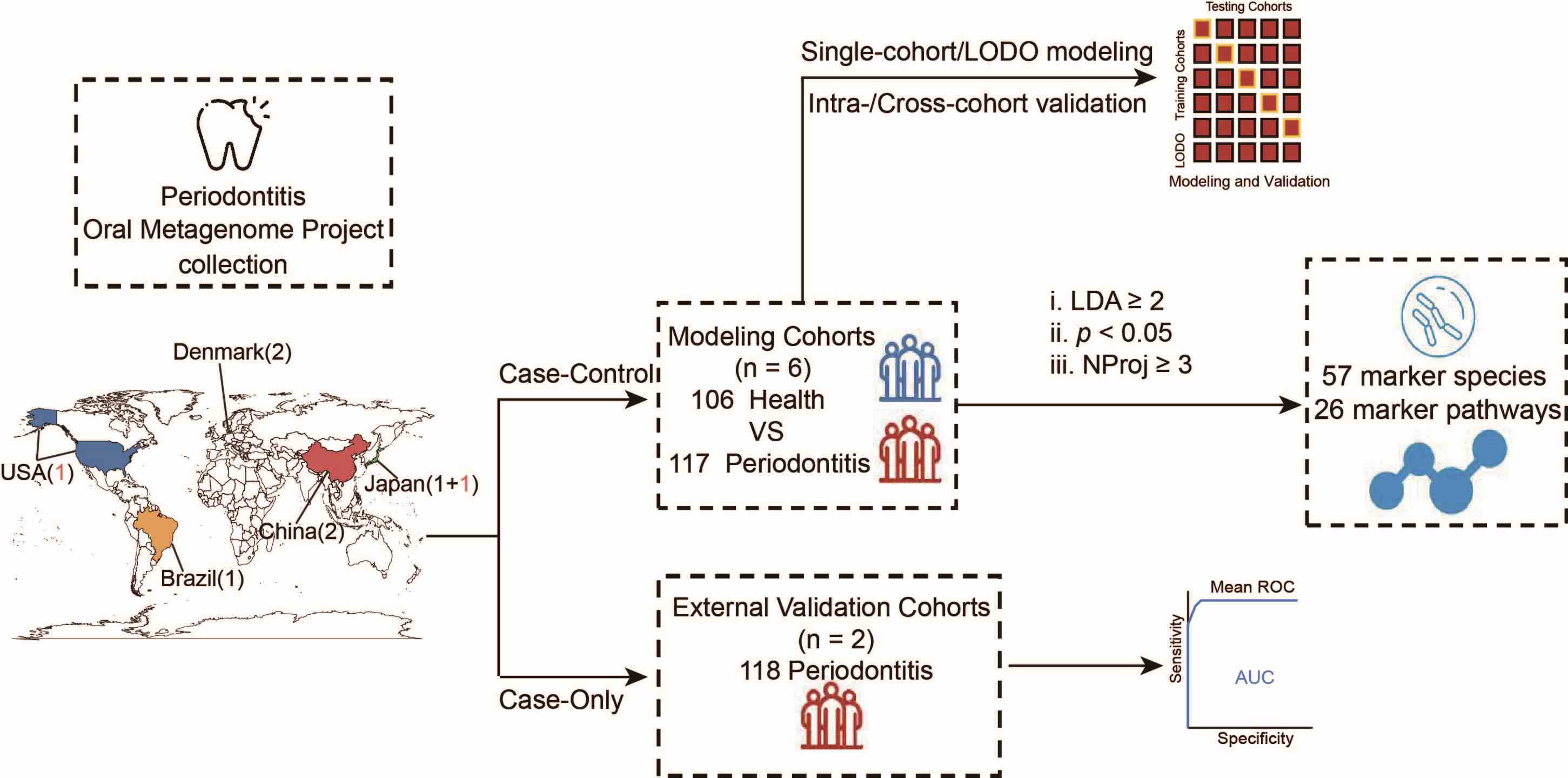
We analyzed eight oral microbiota shotgun metagenomic sequencing cohorts from five countries and three continents, identifying 54 species biomarkers and 26 metabolic biomarkers consistently altered in health and disease states across three or more cohorts. Additionally, machine learning models based on taxonomic profiles achieved high accuracy in distinguishing periodontitis patients from controls (internal and external areas under the receiver operating characteristic curves of 0.86 and 0.85, respectively). These results support metagenome-based diagnosis of periodontitis and provide a foundation for further research and effective treatment strategies.
Probio-M9, a breast milk-originated probiotic, alleviates mastitis and enhances antibiotic efficacy: Insights into the gut–mammary axis
- 09 July 2024
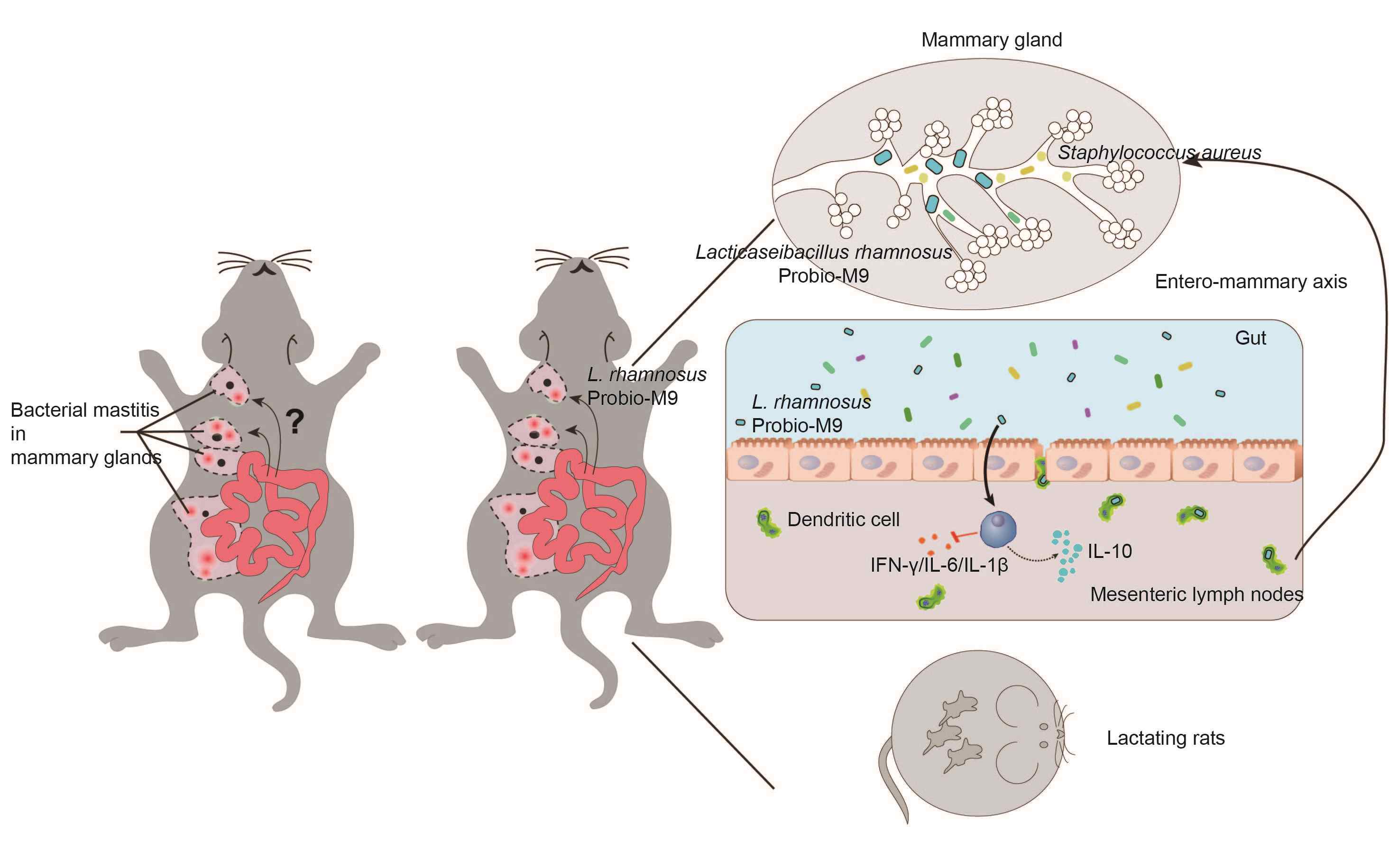
Breast milk naturally contains lactic acid bacteria, but their precise origin remains a subject of debate. In this study, we utilized a rat mastitis animal model to investigate the potential of a breast milk-derived probiotic strain, Lacticaseibacillus rhamnosus Probio-M9, in alleviating mastitis and enhancing the efficacy of antibiotic treatment. Through histopathological analysis of mammary tissue, we observed that Probio-M9 effectively relieved mastitis, mitigated inflammation, and improved the response to antibiotic treatment. Metagenomic analysis further revealed that Probio-M9 enhanced interactions among gut microbes, accompanied by an increase in the relative abundance of Ruminococcaceae and the regulation of specific genes and carbohydrate-active enzymes, subsequently impacting host immunity. Additionally, an intriguing finding was the translocation of live Probio-M9 from the gut to the mammary tissue only during bacterial mastitis and lactation, likely facilitated through lymphatic circulation. These findings advance our understanding of the intricate gut–mammary axis and provide valuable insights into the potential health benefits of probiotic interventions.













