
Synthetic microbial communities: Sandbox and blueprint for soil health enhancement
- 11 February 2024
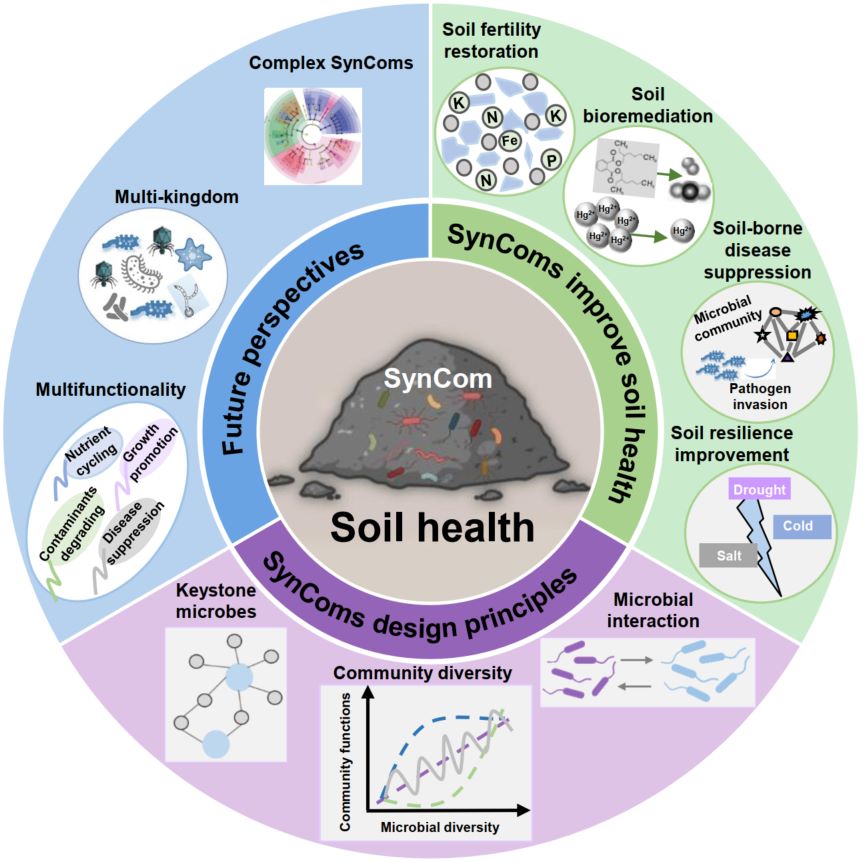
We summarize here the use of SynComs in improving various dimensions of soil health, including fertility, pollutant removal, soil-borne disease suppression, and soil resilience; as well as a set of useful guidelines to assess and understand the principles for designing SynComs to enhance soil health. Finally, we discuss the next stages of SynComs applications, including highly diverse and multikingdom SynComs targeting several functions simultaneously.
ggVennDiagram: Intuitive Venn diagram software extended
- 14 February 2024
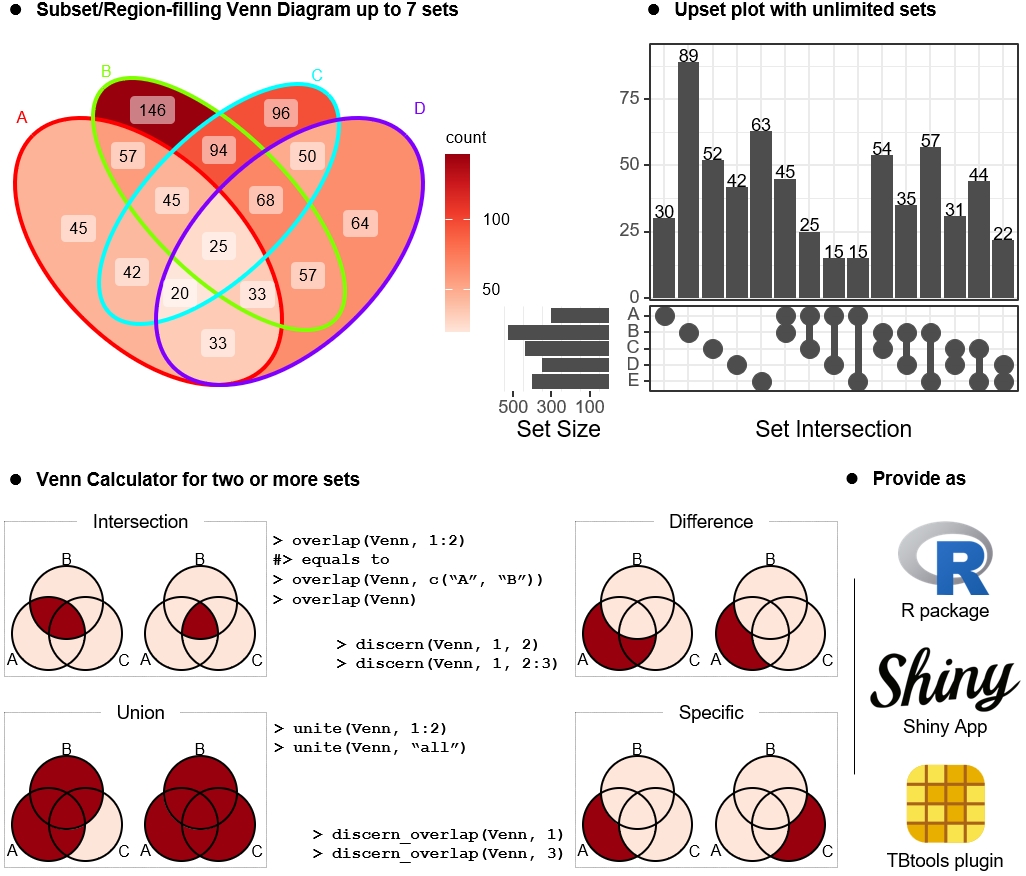
Highlights of ggVennDiagram include: (1) Subset/Region filling Venn diagram up to seven sets; (2) Upset plot with unlimited sets; (3) Venn Calculator for two or more sets; (4) Provide as R package, Shiny App, and TBtools plugin.
Bacterial load in meconium
- 13 February 2024
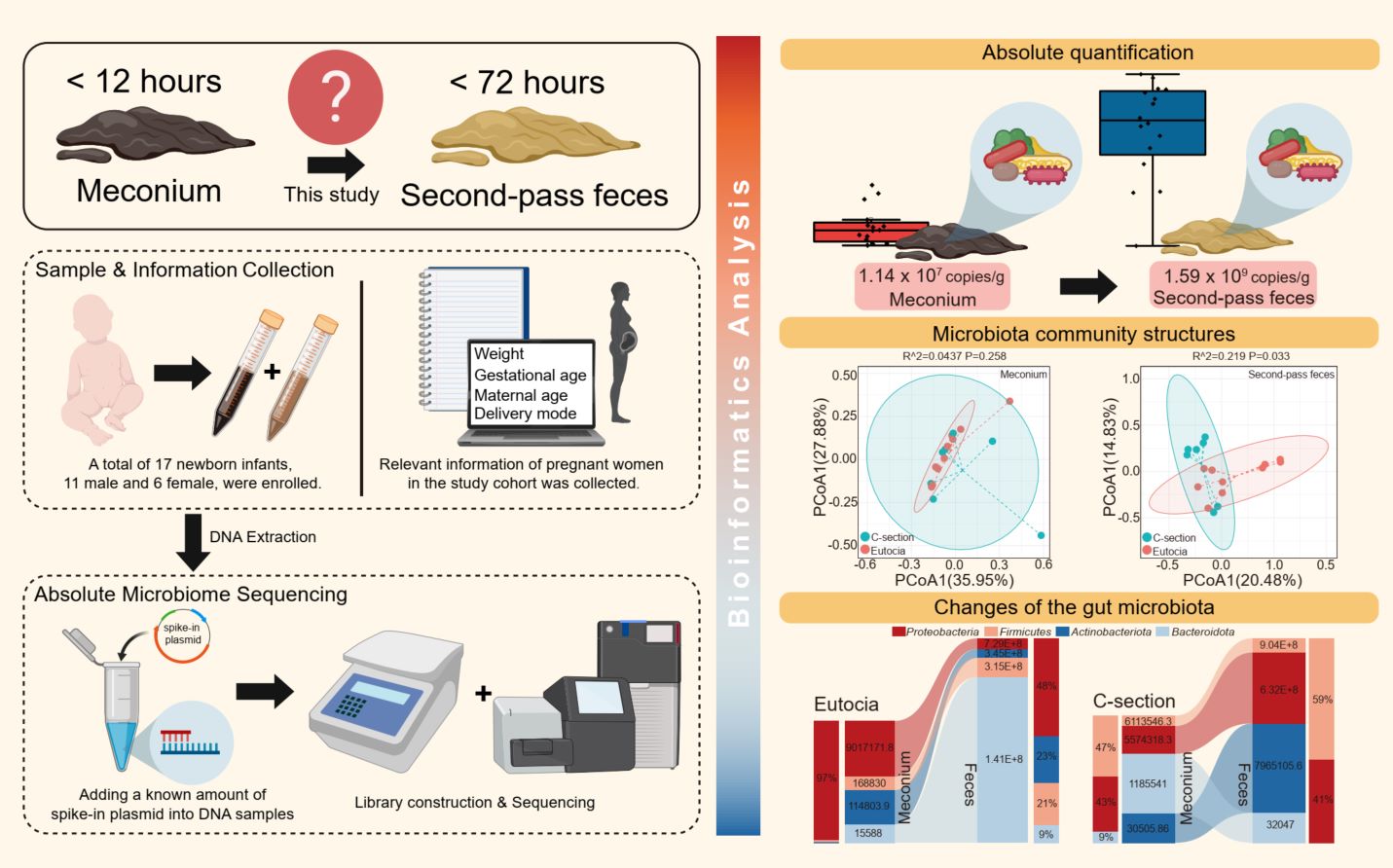
The spike-in plasmid method was utilized to perform an analysis on meconium and second-pass feces, yielding both relative and absolute quantitative results. With the absolute quantitative data, the abundance of bacteria in 17 meconium samples and 17 second-pass fecal samples were found to be 1.14×10^7 and 1.59×10^9 copies/g, respectively. The mode of delivery can significantly influence the alterations and compositions of gut bacteria in a newborn within 72h.
CBD2: A functional biomarker database for colorectal cancer
- 04 December 2023
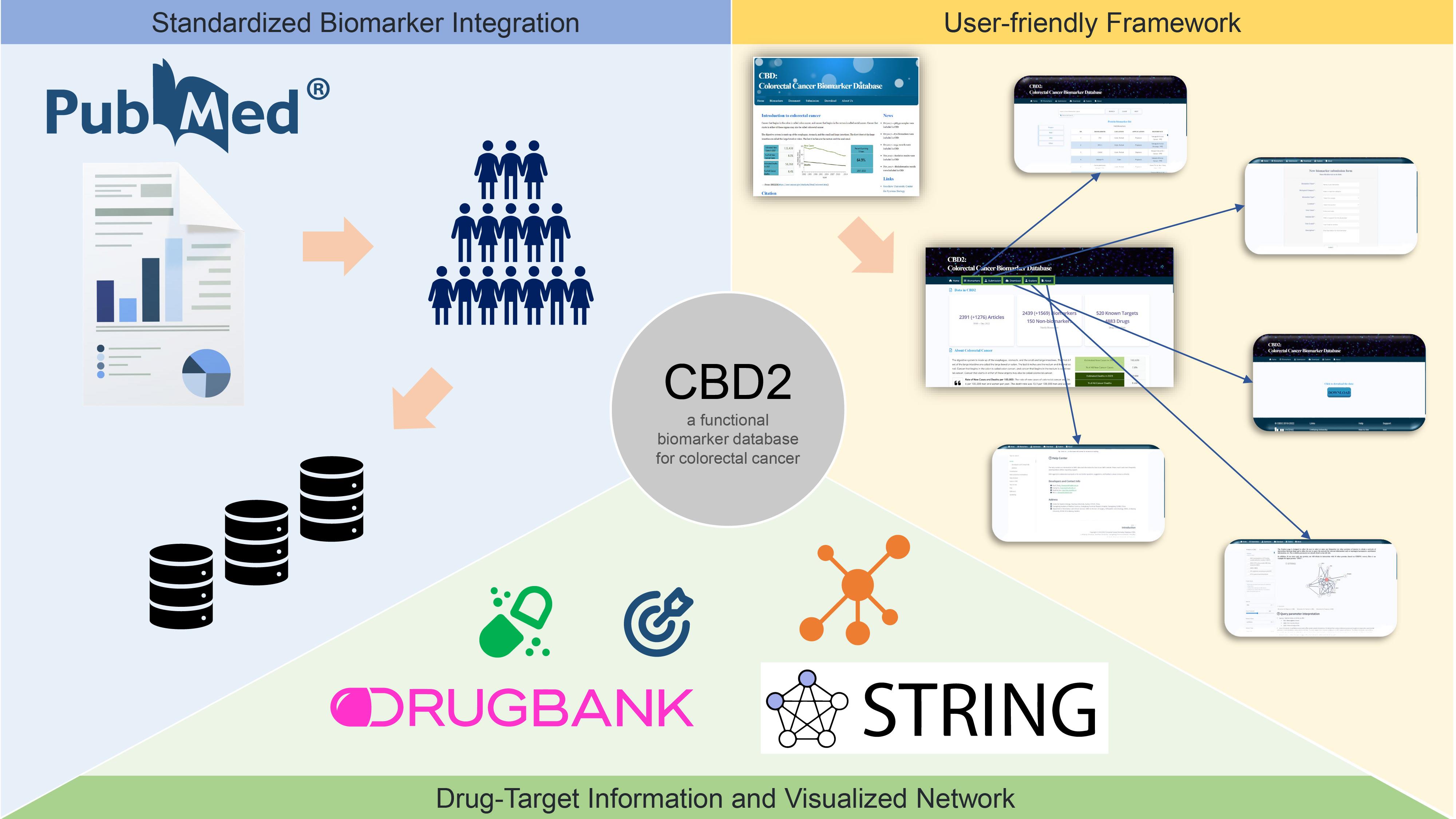
CBD2 integrates biomarkers derived from diverse biological sources and clinical applications, employing a standardized approach for annotation and classification. CBD2 facilitates precision medicine in colorectal cancer by providing abundant data, including drug–target information and visualized network analysis functions. With its user-friendly interface, CBD2 aids in the identification and development of biomarkers for colorectal cancer.
Wekemo Bioincloud: A user-friendly platform for meta-omics data analyses
- 13 February 2024
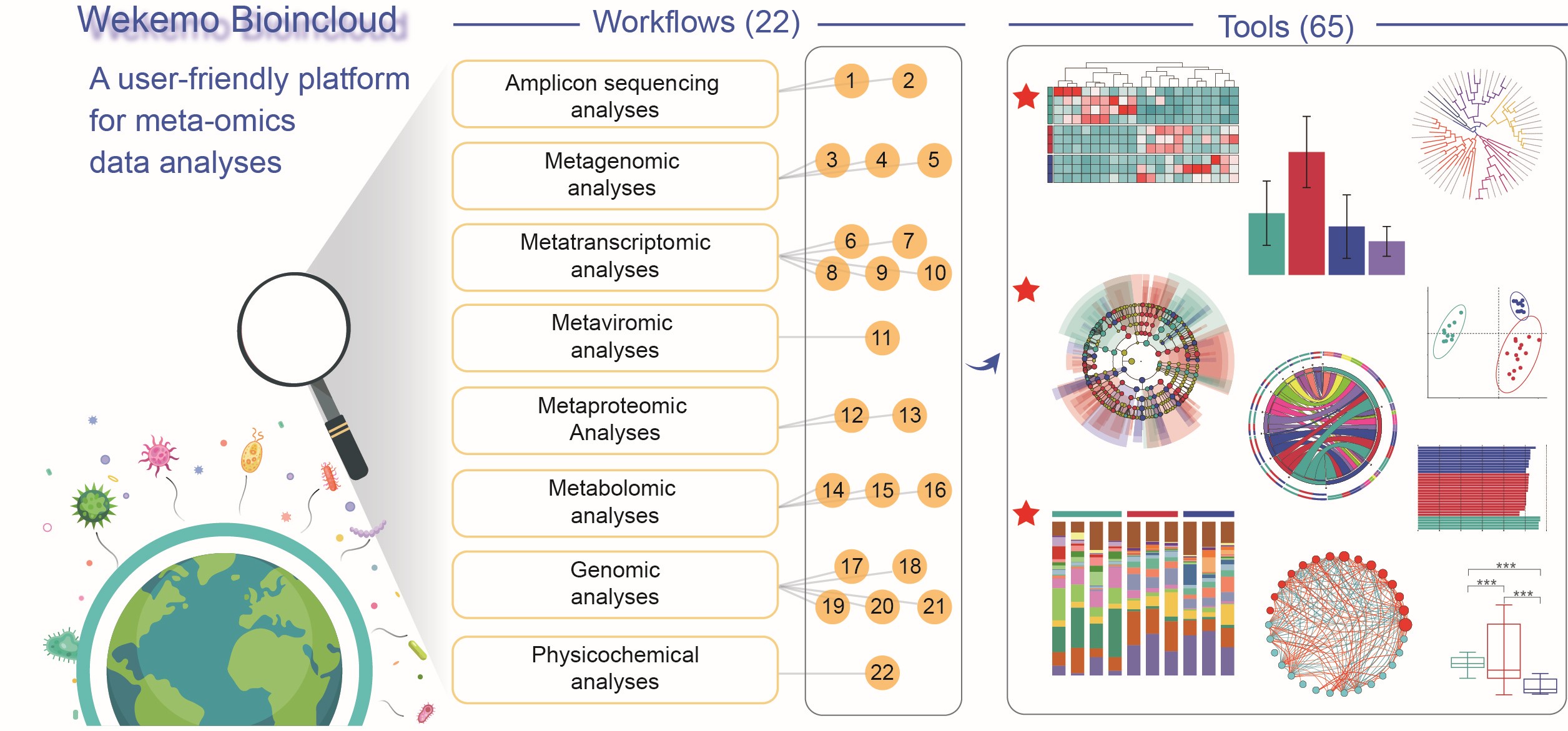
Wekemo Bioincloud is a specialized platform for meta-omics studies that addresses scalability challenges in handling vast microbial community data with 22 workflows and 65 visualization tools. It provides comprehensive analysis, online vector output modification, and ensures privacy with a registration-independent dashboard system.
Duck pan-genome reveals two transposon insertions caused bodyweight enlarging and white plumage phenotype formation during evolution
- 17 December 2023
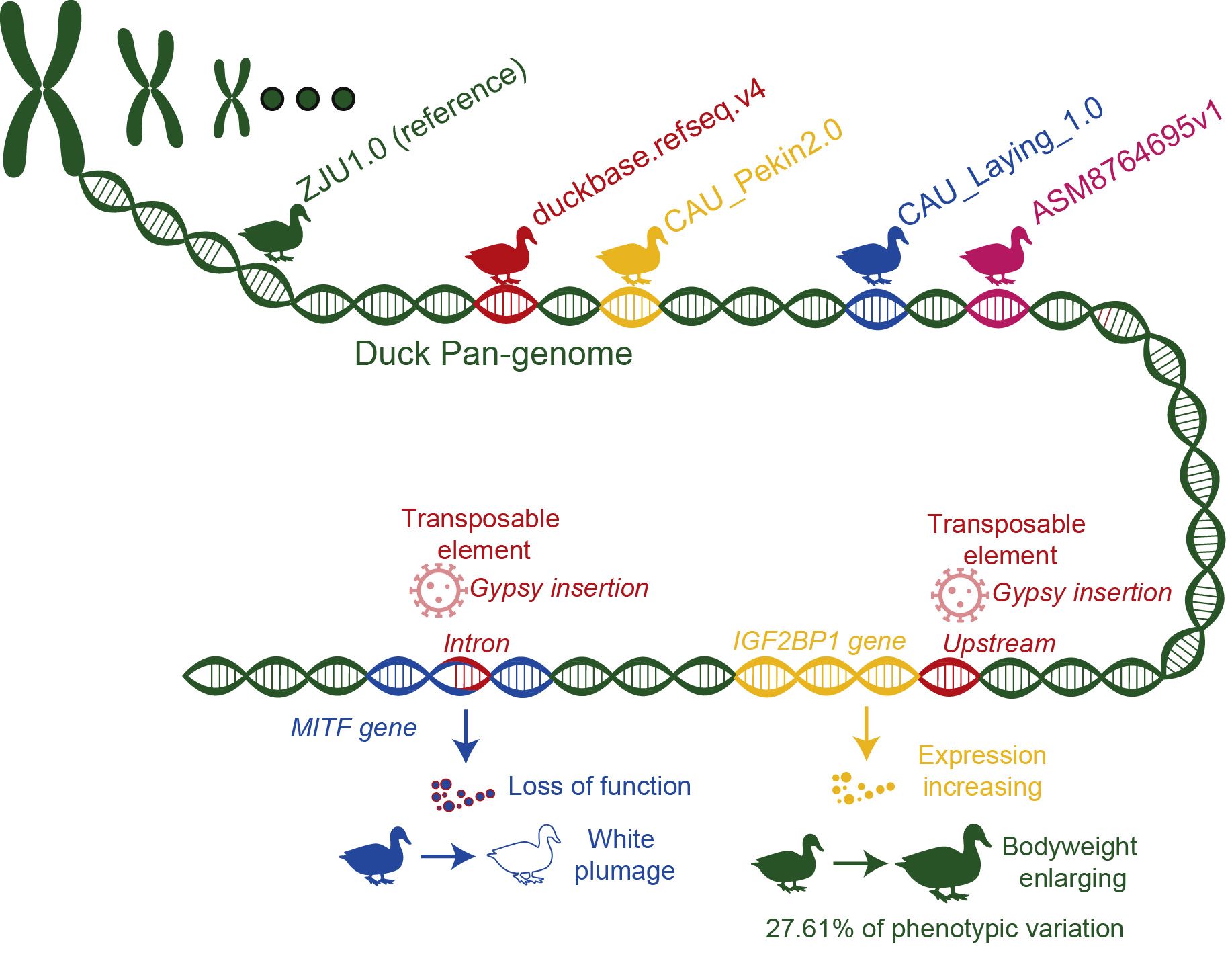
We present the first duck pan-genome constructed using five genome assemblies capturing ∼40.98 Mb new sequences absent from the reference genome. We find a significant portion of the detected structural variants were derived from transposable element (TE) activity. Many of these are located within gene bodies or regulatory regions, potentially linked to duck domestication and enhancement. We used two representative examples to show how TE insertions can lead phenotypic diversity, highlighting IGF2BP1's role in bodyweight and MITF's influence on white plumage in ducks. Notably, the Gypsy insertion in the IGF2BP1 promoter, to our knowledge, explains the largest effect on bodyweight among avian species (27.61% of phenotypic variation).
Black rice diet alleviates colorectal cancer development through modulating tryptophan metabolism and activating AHR pathway
- 15 January 2024
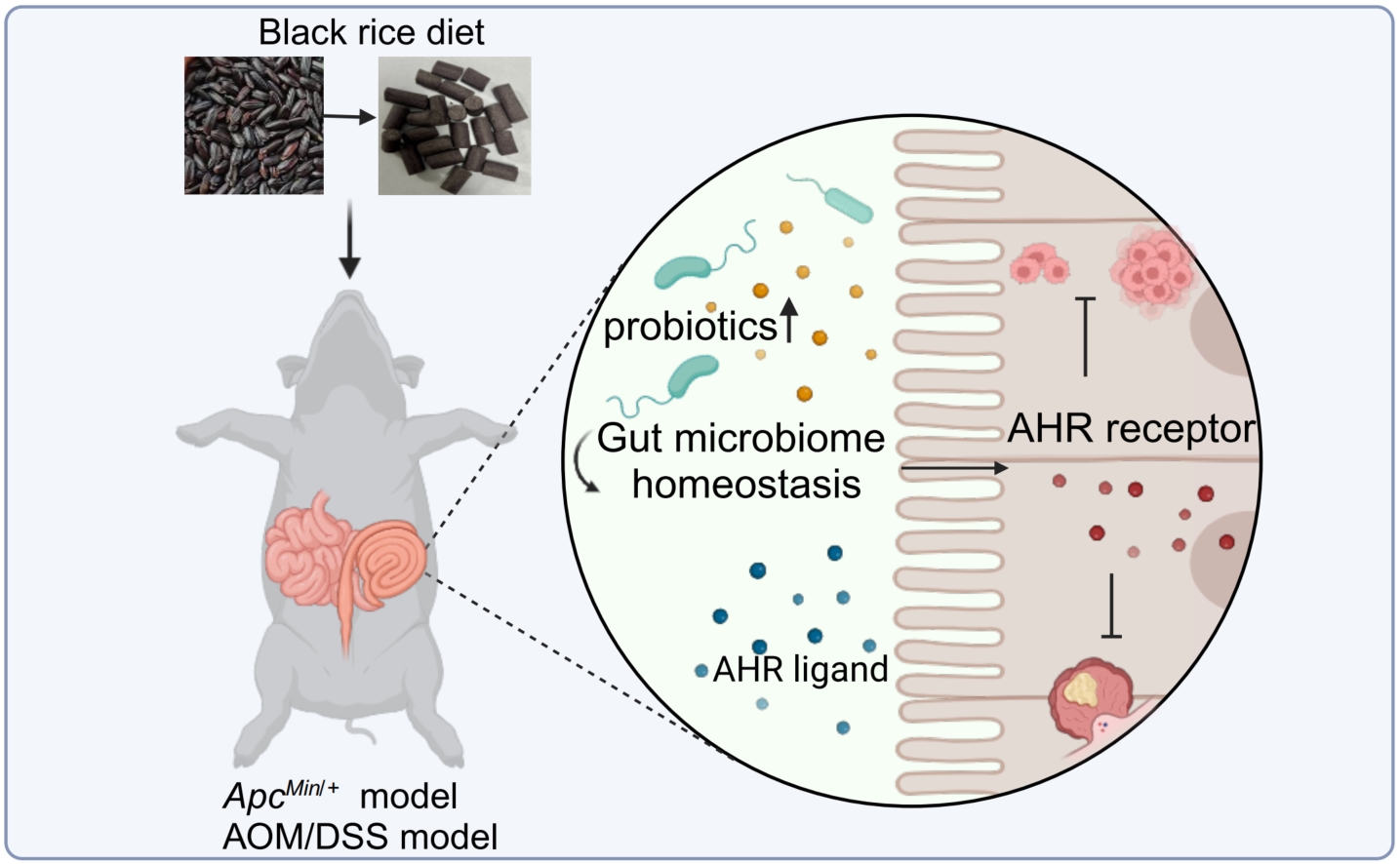
Black rice diet can regulate gut microbial homeostasis in the colorectal cancer (CRC) mice model, enhance the abundance of beneficial bacteria, create aromatic hydrocarbon receptor ligand metabolites, activate the intestinal aryl hydrocarbon receptor (AHR) pathway, and suppress the development of CRC.
Clostridium butyricum and carbohydrate active enzymes contribute to the reduced fat deposition in pigs
- 03 January 2024
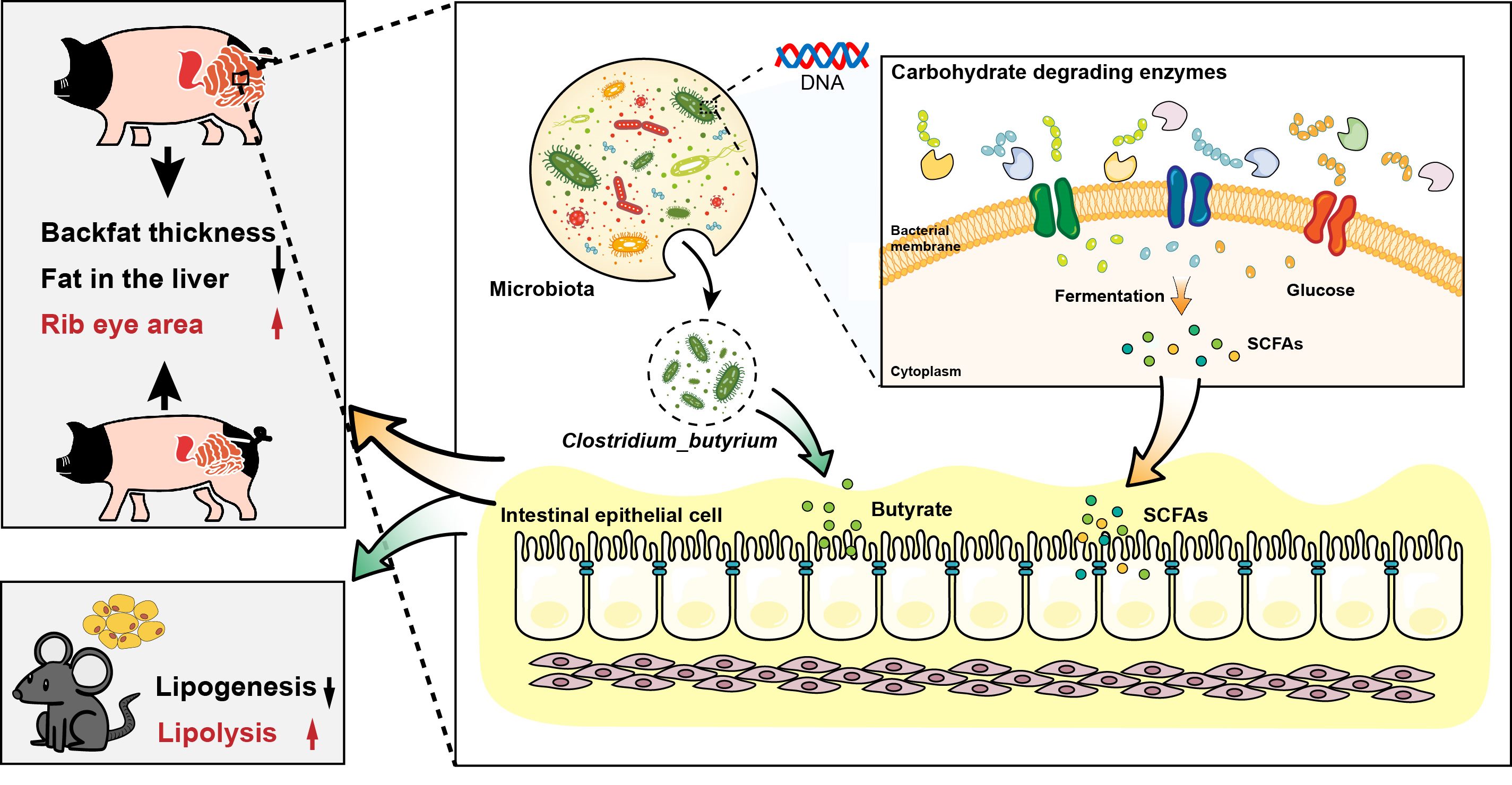
In the present study, we found significant differences in microbial composition and potential functional capacity among different gut locations in Jinhua pigs with distinct fatness phenotypes. Second, we identified that Jinhua pigs with lower fatness exhibited higher levels of short-chain fatty acids in the colon, highlighting their enhanced carbohydrate fermentation capacity. Notably, Clostridium butyricum might be a representative bacterial species from Jinhua pigs with lower fatness, and a significantly higher percentage of its genome was dedicated to CAZyme glycoside hydrolase family 13 (GH13). Finally, a subsequent mouse intervention study substantiated the beneficial effects of C. butyricum isolated from experimental pigs, suggesting that it may possess characteristics that promote the utilization of carbohydrates and hinder fat accumulation. Remarkably, when Jinhua pigs were administered C. butyricum, similar alterations in the gut microbiome and host fatness traits were observed, further supporting the potential role of C. butyricum in modulating fatness.
Daily occupational exposure in swine farm alters human skin microbiota and antibiotic resistome
- 01 January 2024
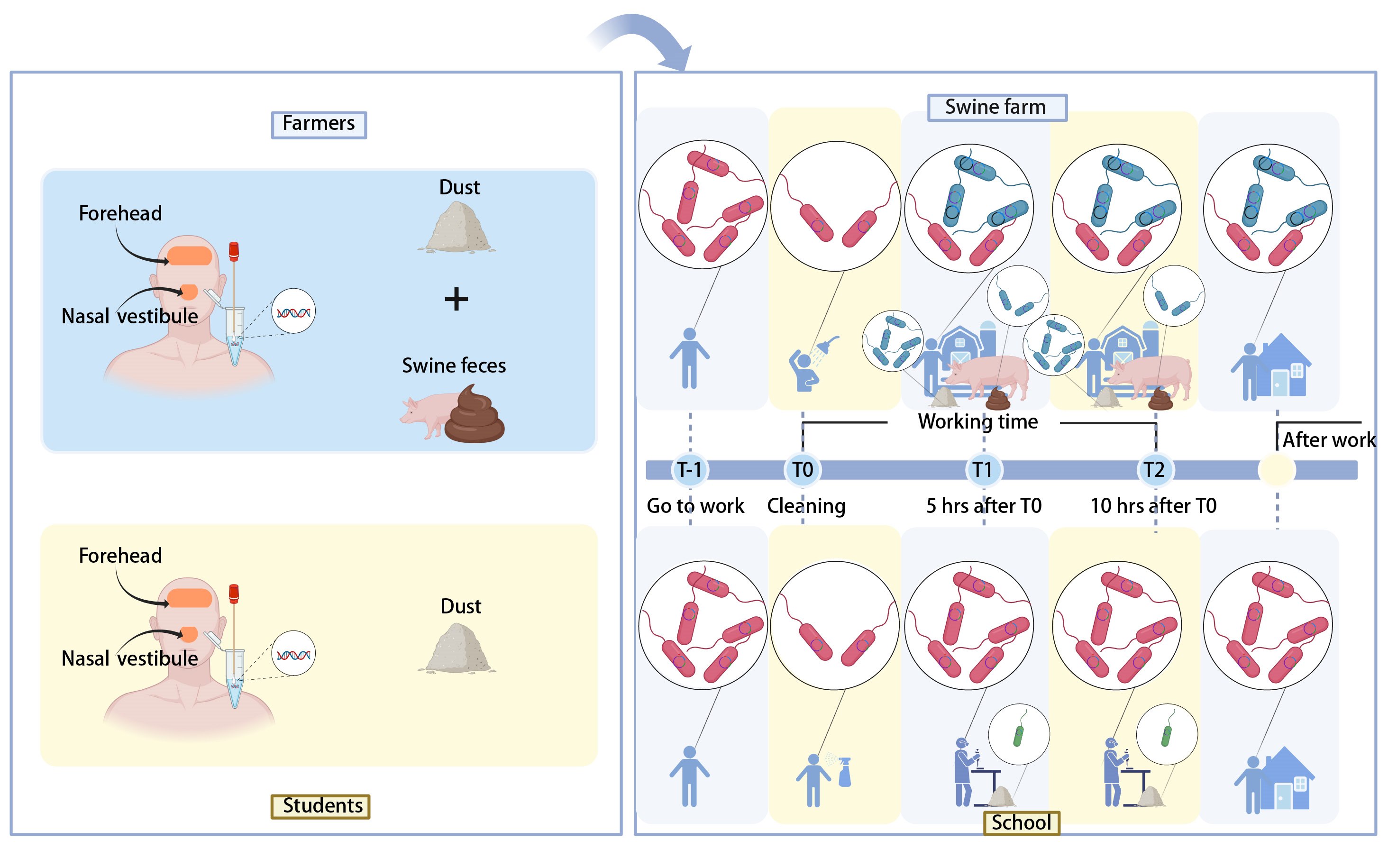
We show that occupational exposure in a livestock environment dynamically reshaped the skin microbiome and resistome. This remodeling effect can take place within hours and the acquired pathogens and antibiotic resistance genes (ARGs) have the potential to expand to the general population using farm workers as an ARG vector. Farm workers deserve special consideration under the One Health framework to curb spreading antibiotic resistance.
Soil enzyme profile analysis for indicating decomposer micro-food web
- 02 January 2024
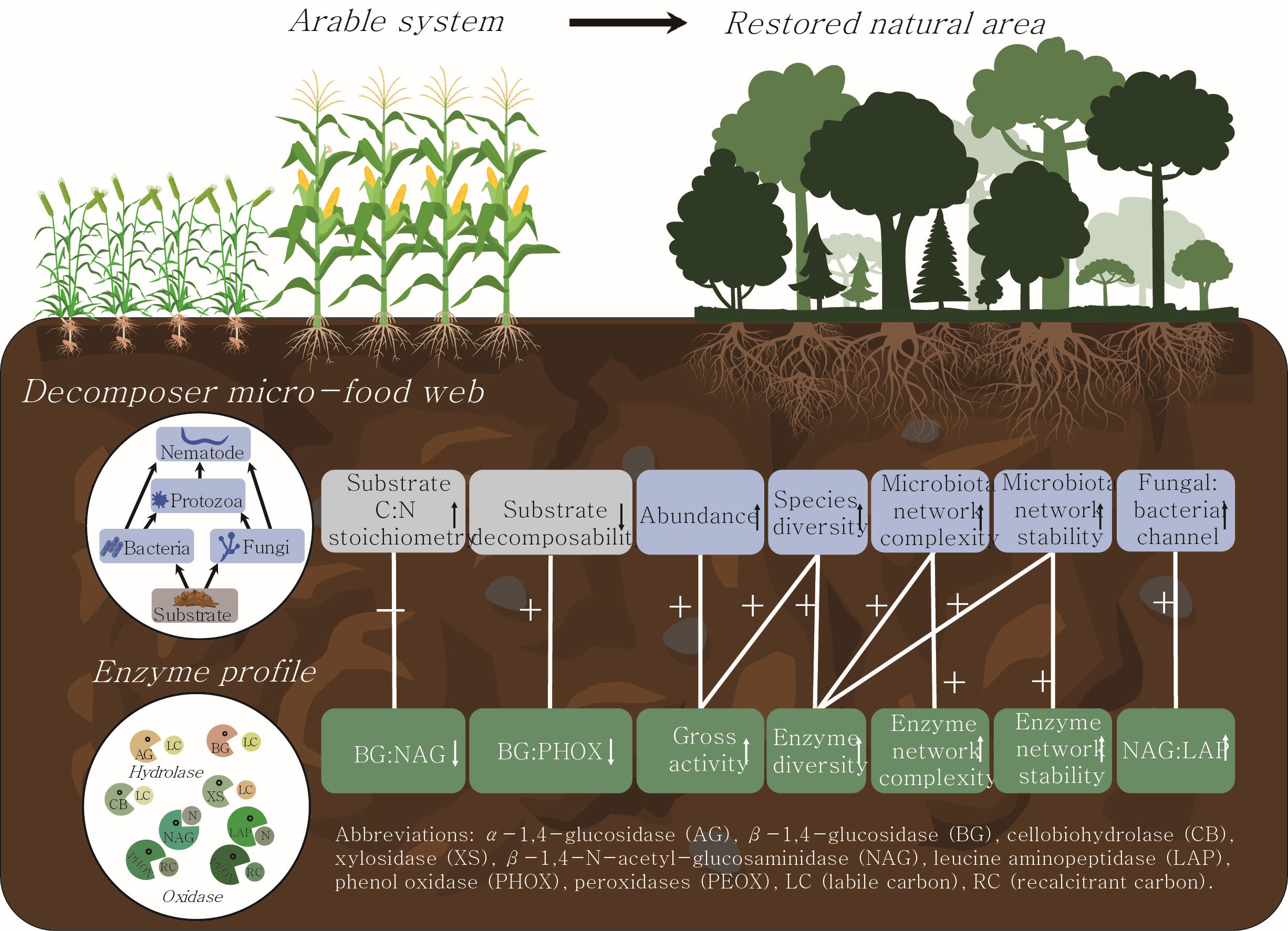
This study proposed the approach of a “soil enzyme profile analysis” whereby a series of enzyme profile indices were hypothesized to comprehensively reflect the micro-food web features. Using a case study on the restored natural area of a post-arable system, we systematically evaluated the shifts in enzyme profile indices in relation to the micro-food web features. +, −, ↑, and ↓ represent positive correlation, negative correlation, increase, and decrease trends, respectively.
From mechanism to application: Decrypting light-regulated denitrifying microbiome through geometric deep learning
- 06 January 2024
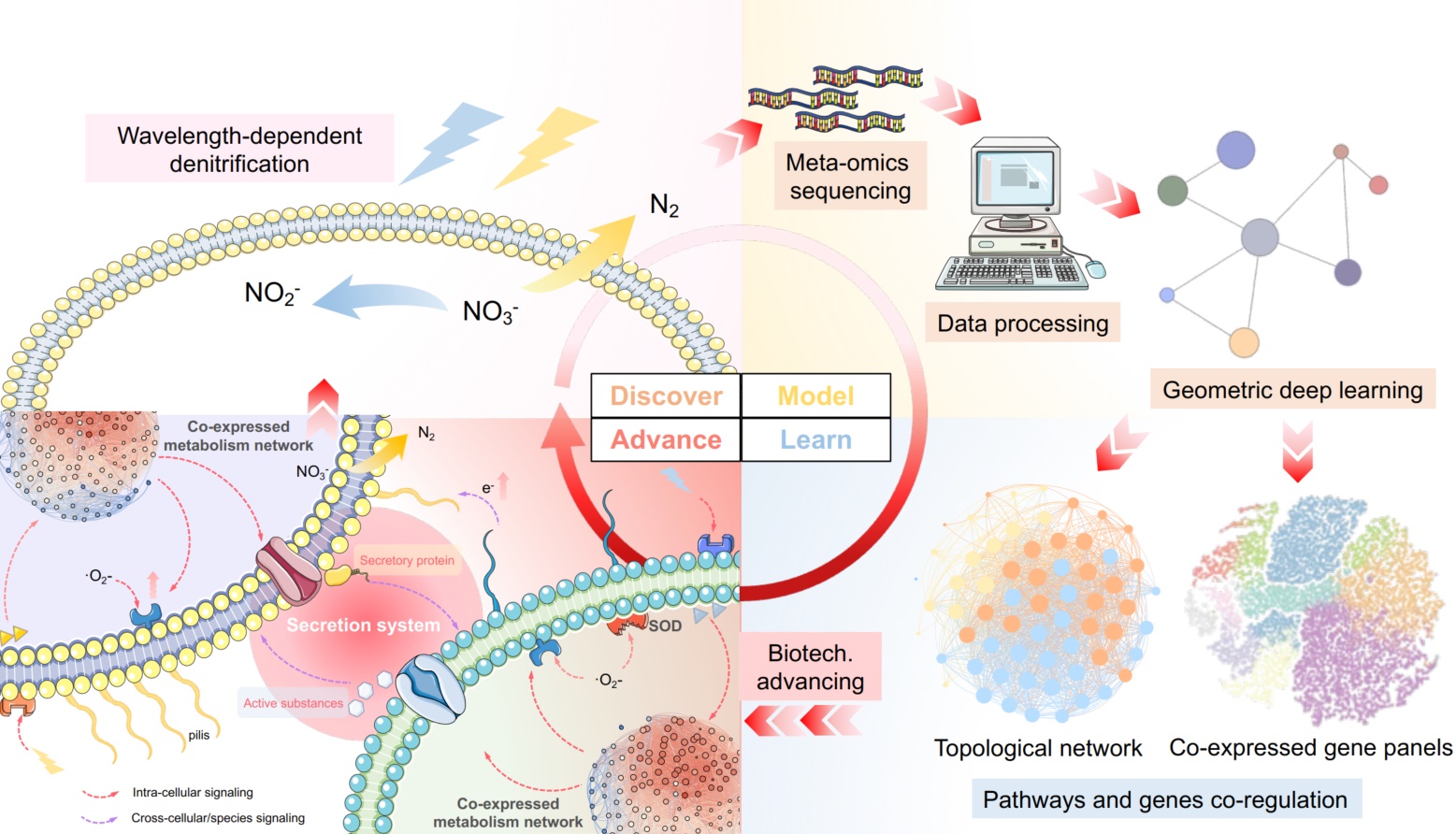
This study proposed and showcased the discover–model–learn–advance (DMLA) cycle for unlocking nature as codebases to empower biological mechanisms decryption and biotechnology development. At the discovering stage, we found the bidirectionally light-regulated denitrification. Then, metatranscriptomic was utilized for geometric deep learning to obtain coexpressed gene panels. On the basis of that, we developed toolkits to learn underlying mechanisms and advance biotechnology, which were validated in the wet lab. Deepening recognition can further drive continuous DMLA cycles to accelerate scientific and biotechnology development.
Glucomannan promotes Bacteroides ovatus to improve intestinal barrier function and ameliorate insulin resistance
- 03 January 2024
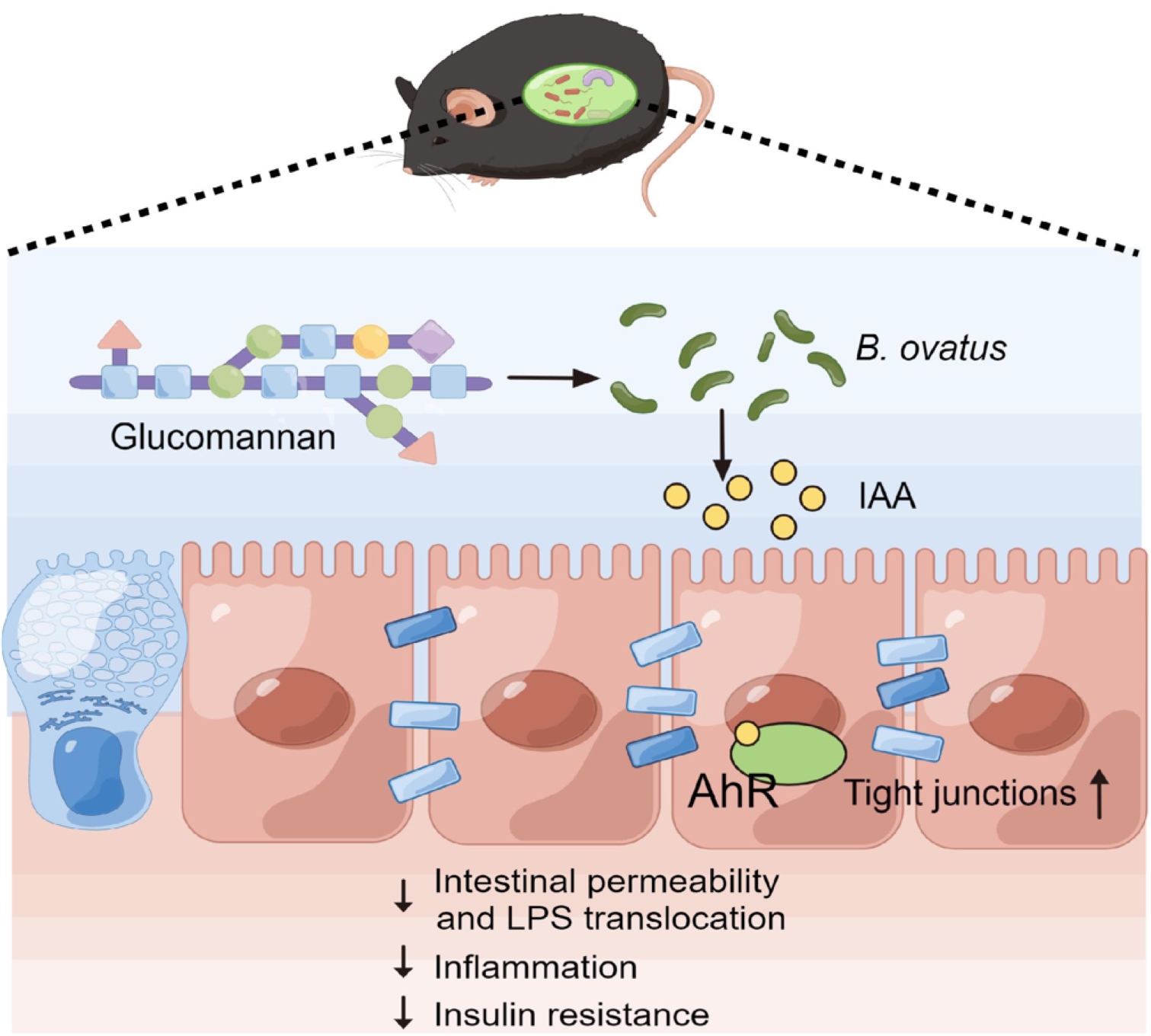
Supplementation with glucomannan alleviates insulin resistance. Glucomannan promotes the growth of Bacteroides ovatus, accompanied by improved intestinal barrier integrity and reduced systemic inflammation. B. ovatus-derived indoleacetic acid is a key bioactive metabolite that fortifies intestinal barrier function via the activation of intestinal aryl hydrocarbon receptor.
Gut microbiota reshapes cancer immunotherapy efficacy: Mechanisms and therapeutic strategies
- 01 January 2024
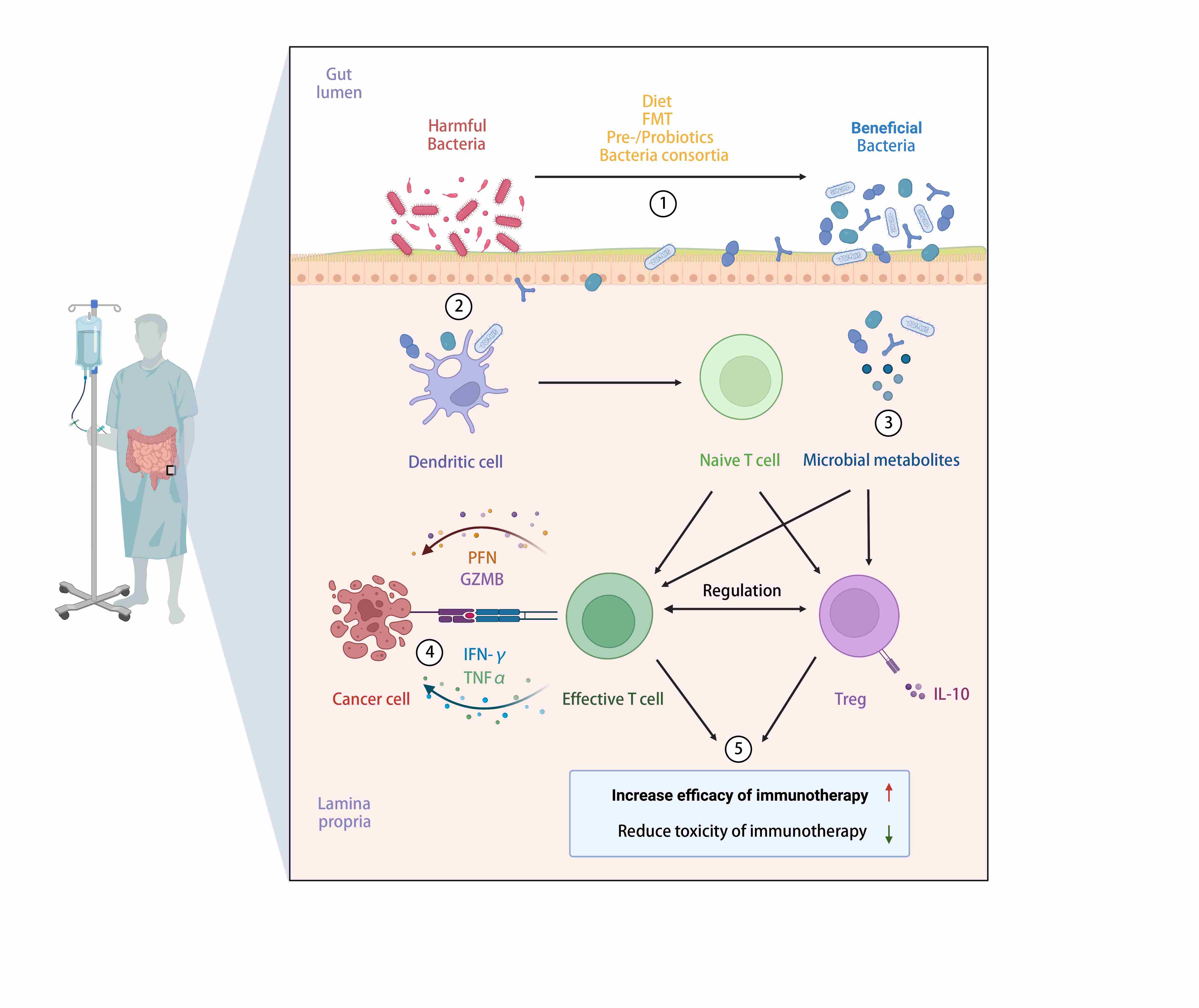
The role of gut microbiota in cancer immunotherapy. Through manipulation of commensals in cancer patients by diet interventions, fecal microbial transplant, prebiotics, probiotics and bacteria consortia, host antitumor immunity can be enhanced by dominance of “beneficial” bacteria in gut lumen and their metabolites. Increased effector T cells and induction of regulatory T cells (Tregs) can be seen in gut-associated lymphoid tissue, which leads to improved clinical outcomes of cancer immunotherapy with lower incidence of immune-related adverse events (fecal microbiota transplant; perforin; granzyme B; Treg).
Recent advances in the characterization of essential genes and development of a database of essential genes
- 02 January 2024
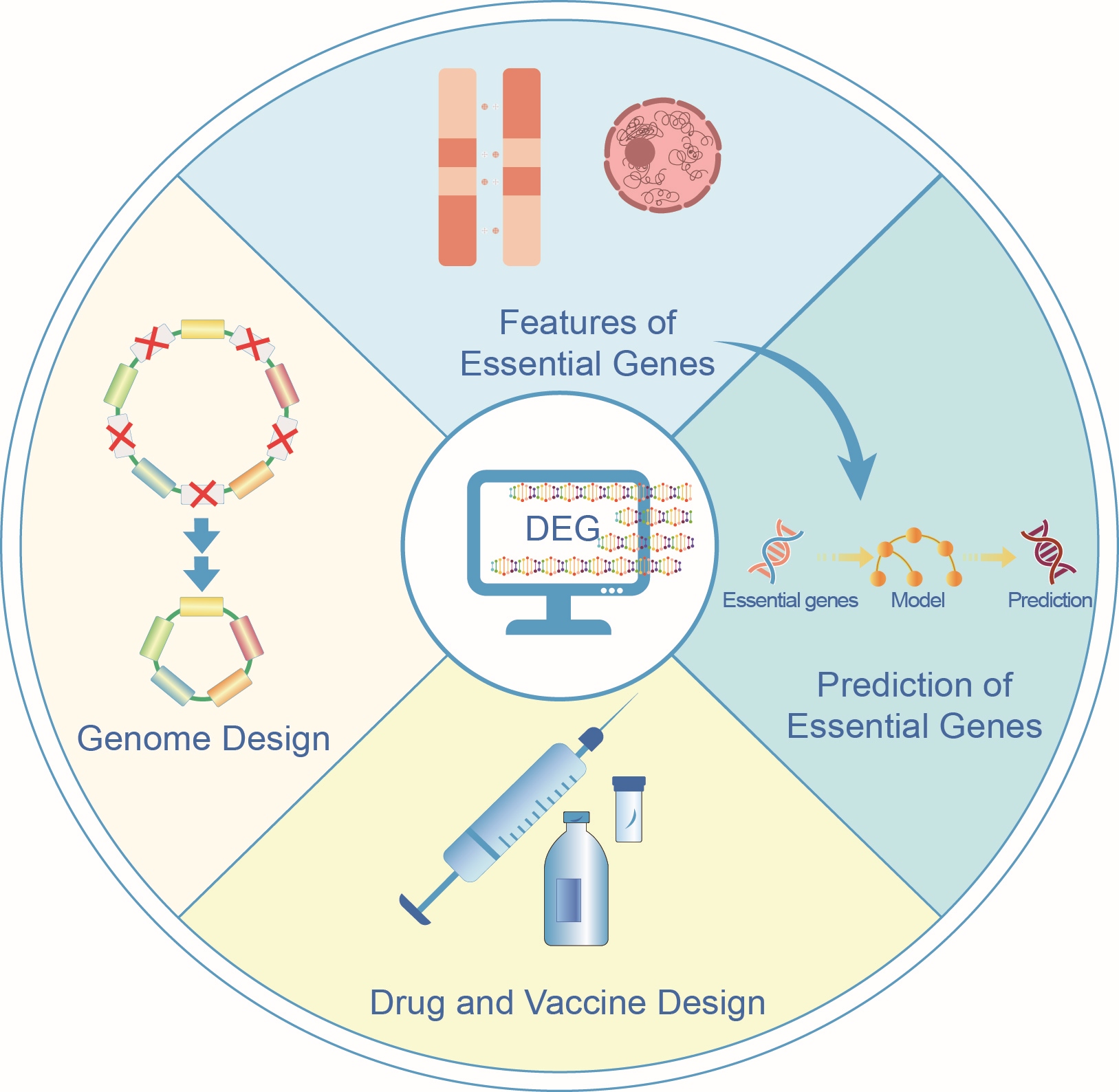
Essential genes are indispensable for the survival and development of organisms, and have been successfully identified using various experimental methods across diverse organisms. DEG, a database of essential genes is dedicated to collecting experimental results on essential genes and has become one of the most commonly used tools for studying essential genes, where the information on essential genes for a specific species can be quickly accessed, with a broad range of applications, such as essential gene feature analysis and prediction, drug and vaccine design, as well as artificial genome design and construction. The definition of essential genes is environment-specific rather than simple binary, which should not be overlooked in the study of essential genes.
A comprehensive analysis of antibiotic resistance genes in the giant panda gut
- 06 February 2024
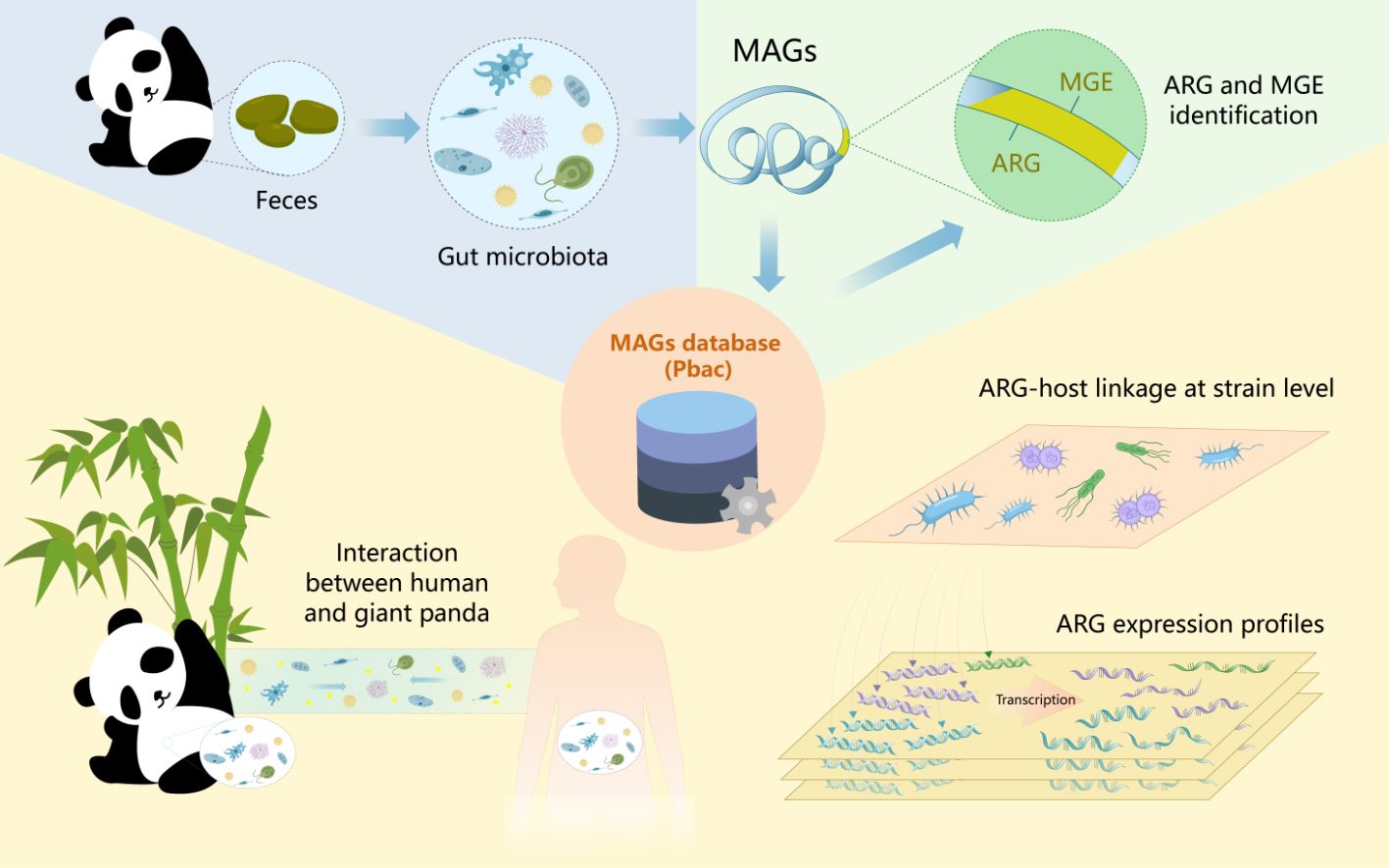
In this study, we have successfully constructed a comprehensive database of metagenome-assembled genomes (MAGs) pertaining to the gut microbiota of the giant panda. Through our analysis, we have identified significant reservoirs of antibiotic resistance genes (ARGs), namely Escherichia coli, Citrobacter portucalensis, and Klebsiella pneumoniae. Furthermore, we have elucidated the primary contributors to ARGs, including Streptococcus alactolyticus and Clostridium SGBP116, in both captive and wild pandas. Additionally, our findings have demonstrated a higher prevalence of ARGs in the metagenome, with notable expression of the RPOB2 gene in S. alactolyticus. Crucially, 1217 ARGs shared homology with human gut ARGs, underscoring the interaction relationship between pandas and human microbiomes. These findings are instrumental in understanding the antibiotic resistance landscape in the giant panda's gut, providing a framework for developing strategies to combat antibiotic resistance and safeguard the health of this endangered species.
PHGD: An integrative and user-friendly database for plant hormone-related genes
- 08 January 2024
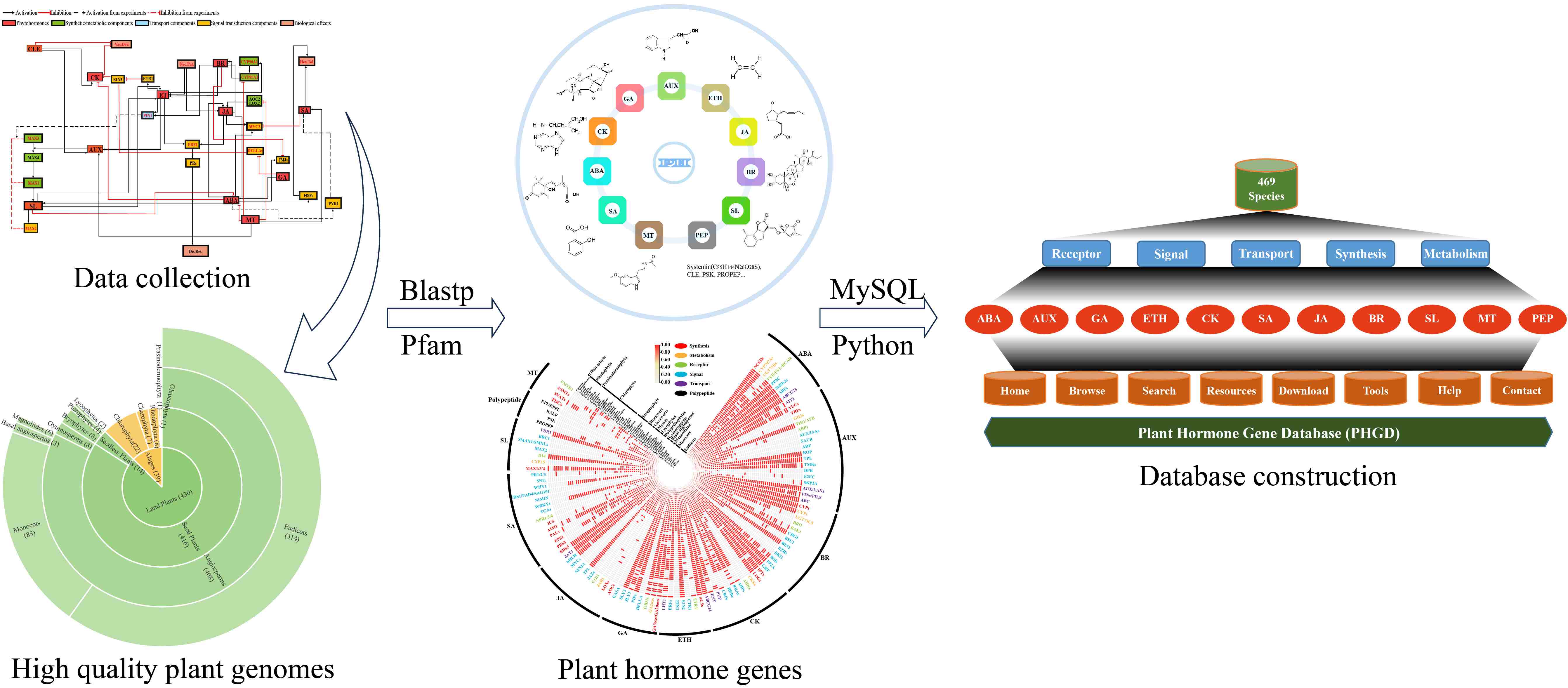
Plant Hormone Gene Database (PHGD) database platform construction pipeline. First, we collected all reported hormone-related genes in the model plant Arabidopsis thaliana, and combined with the existing experimental background, mapped the hormone–gene interaction network to provide a blueprint. Next, we collected 469 high-quality plant genomes. Then, bioinformatics was used to identify hormone-related genes in these plants. Finally, these genetic data were programmed to be stored in a database and a platform website PHGD was built. PHGD was divided into eight modules, namely Home, Browse, Search, Resources, Download, Tools, Help, and Contact. We provided data resources and platform services to facilitate the study of plant hormones.










