
SeqKit2: A Swiss army knife for sequence and alignment processing
- 05 April 2024
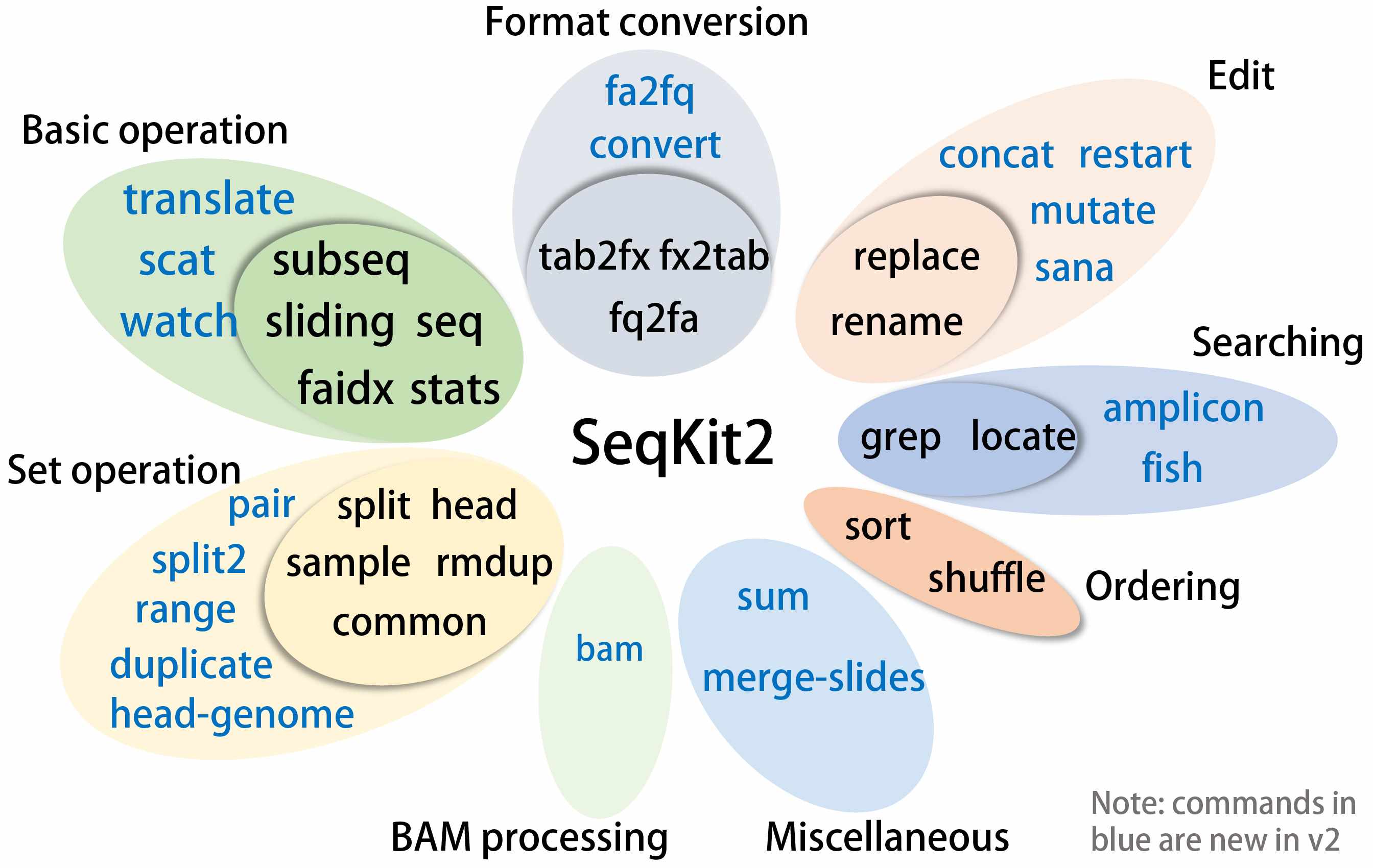
SeqKit2 has significantly expanded its capabilities, doubling the number of subcommands from 19 to 38, and adding support for three more compression file formats. This enhances its versatility and efficiency in sequence and alignment processing. Benchmarking results demonstrate that SeqKit2 consistently outperforms its predecessor and remains competitive with other similar tools. In addition, SeqKit2 has notably improved user-friendliness with the introduction of new features, such as autocompletion for subcommands and command-line flags and enhanced error-handling mechanisms.
Visualizing set relationships: EVenn's comprehensive approach to Venn diagrams
- 11 April 2024
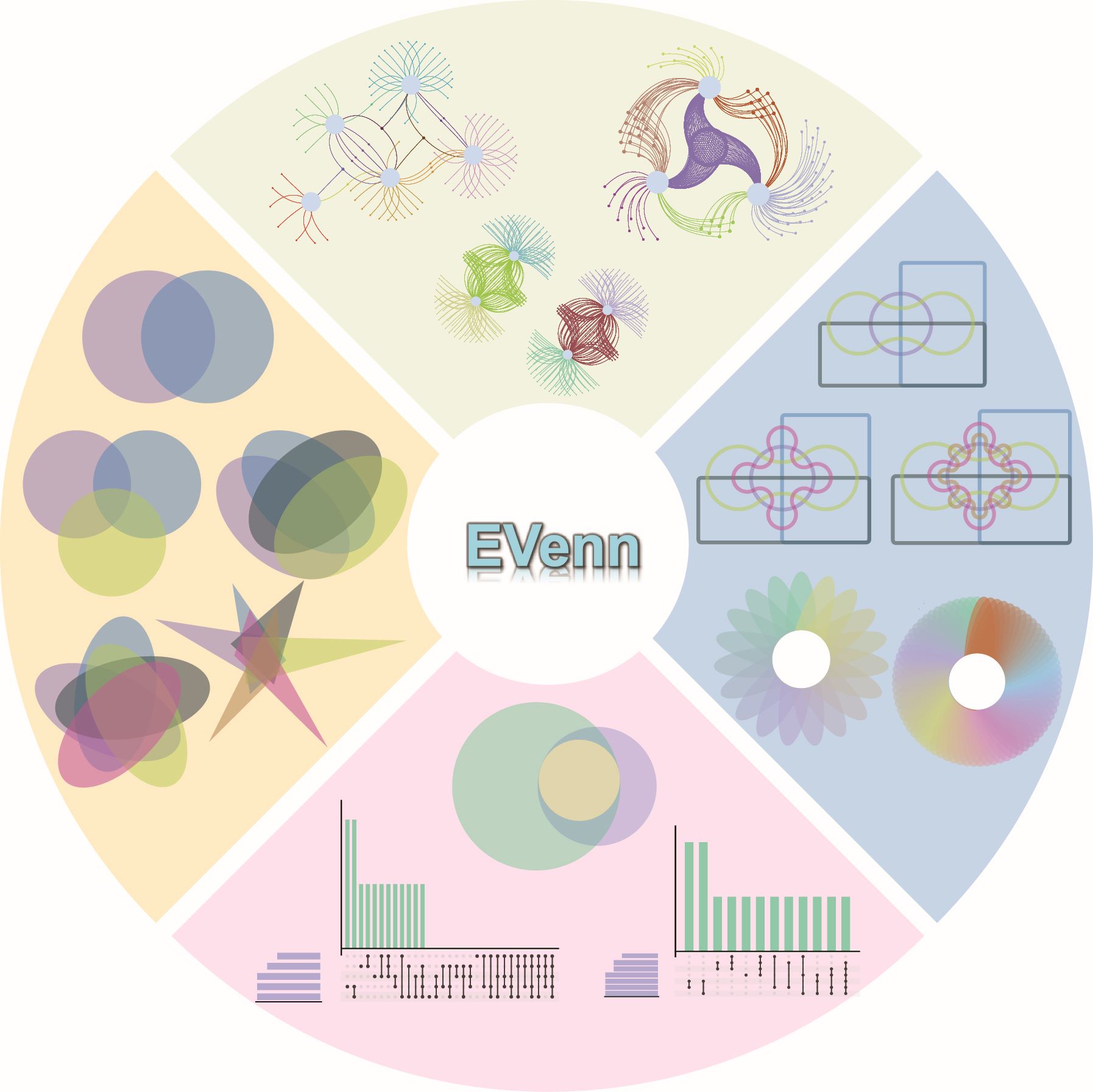
The use of Venn diagrams greatly aids in illustrating and visualizing set relationships within metabolomics, genomics, transcriptomics, and proteomics. Here, we introduce EVenn, an all-encompassing platform that provides a unified interface for a wide range of Venn diagram functionalities. This protocol delineates the features of EVenn, encompassing interactive Venn diagrams, interactive Edwards diagrams, Euler diagrams, UpSet plots, flower plots, and Venn network diagrams, solidifying its role as an invaluable tool for the analysis of multiomics data.
Bacmethy: A novel and convenient tool for investigating bacterial DNA methylation pattern and their transcriptional regulation effects
- 19 March 2024
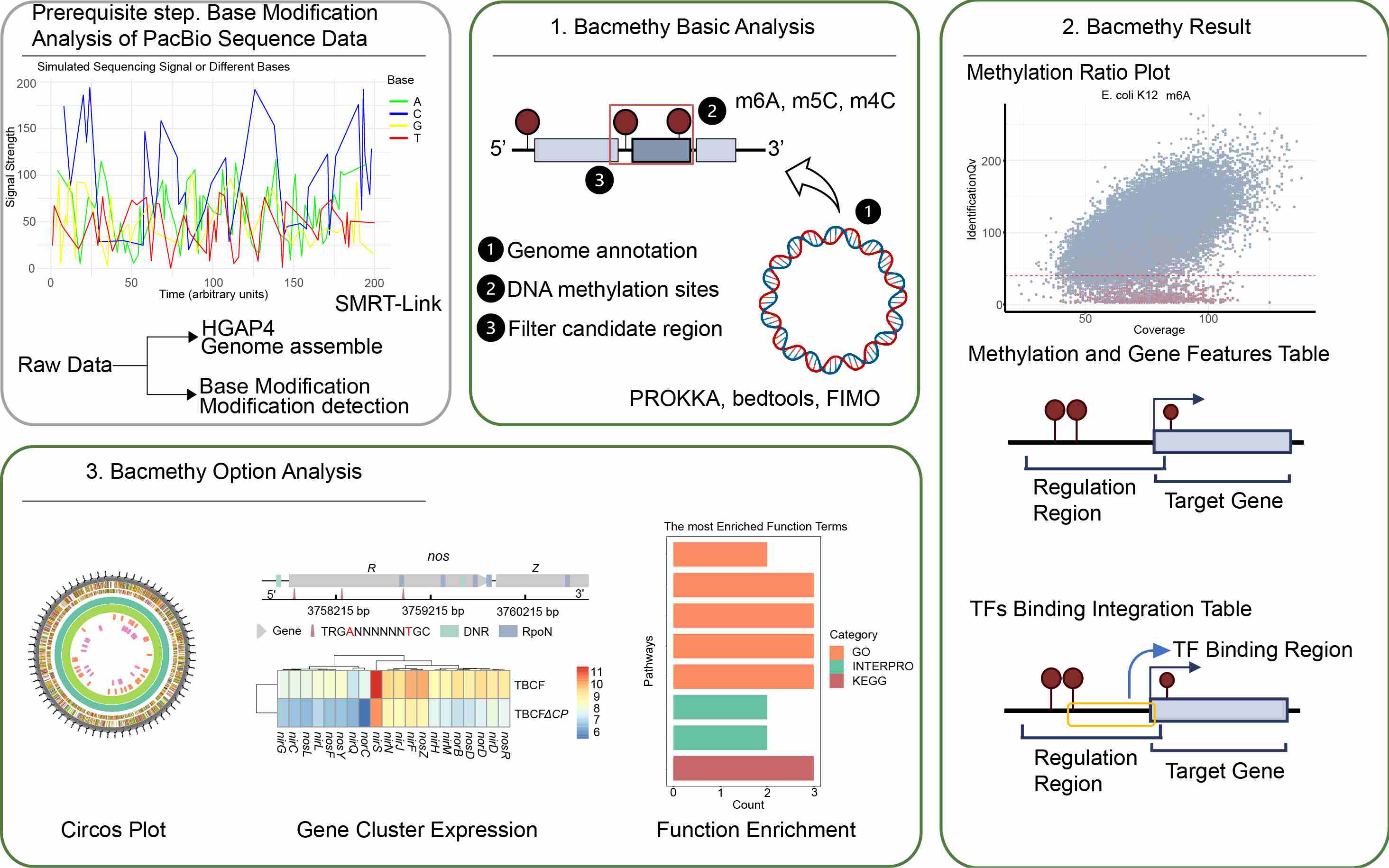
Bacmethy tool provides a one-stop analysis and visualization pipeline for effectively characterizing bacterial DNA methylation modification features and predicting the regulation patterns. Bacmethy offers both a local run function and an online interface analysis service, providing significant convenience for researchers without coding abilities. Bacmethy provides useful information for decoding the underlying molecular mechanisms of how DNA methylation regulates bacterial cellular and physiological functions.
A panoramic view of the virosphere in three wastewater treatment plants by integrating viral-like particle-concentrated and traditional non-concentrated metagenomic approaches
- 29 March 2024
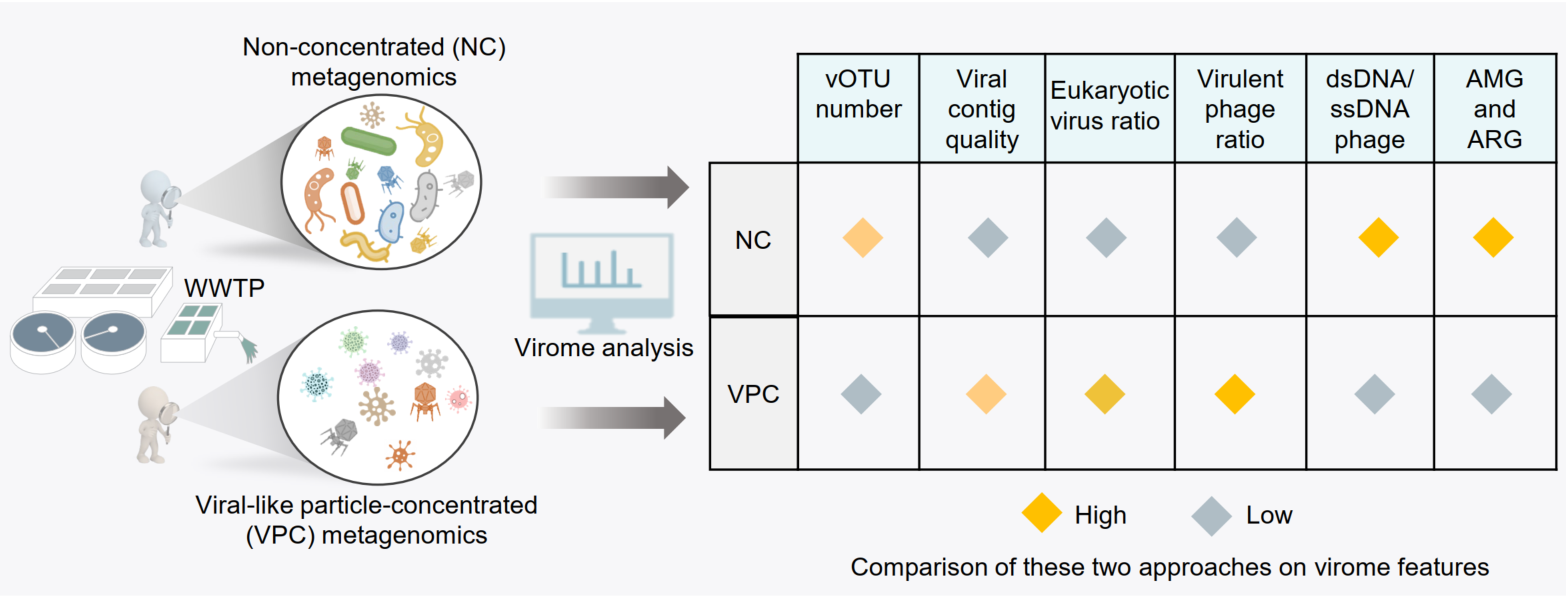
Our comparative analysis unveiled the distinct advantages of two methods in capturing the complexity of viral communities and their functions in wastewater treatment plants. The non-concentrated (NC) metagenomic approach identified a higher number of viral contigs, though it had a lower yield of high-quality contigs. In contrast, the viral-like particle-concentrated (VPC) metagenomic method outperformed in recovering a larger proportion of high-quality viral contigs, and significantly enriched free eukaryotic viruses. Notably, the phage-derived auxiliary metabolic genes and antibiotic resistance genes were predominantly located in the NC metagenomes rather than the VPC metagenomes. Overall, the integration of both VPC and NC metagenomic approaches offers a robust framework for a thorough and nuanced exploration of viral communities in wastewater environments, contributing valuable insights for the advancement of wastewater treatment technologies.
Unraveling interindividual variation of trimethylamine N-oxide and its precursors at the population level
- 30 March 2024
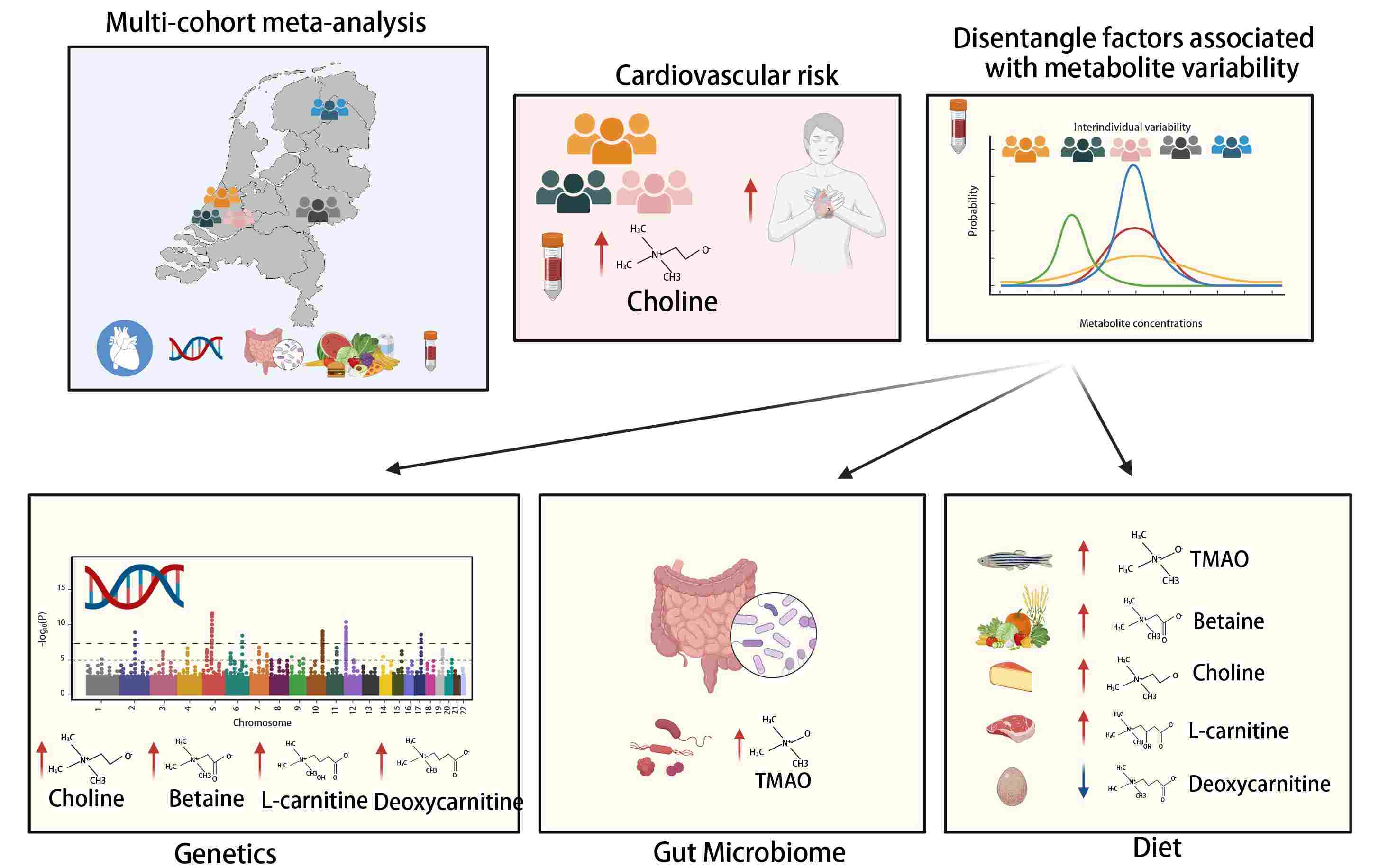
Trimethylamine N-oxide (TMAO) is a circulating microbiome-derived metabolite implicated in the development of atherosclerosis and cardiovascular disease (CVD). We investigated whether plasma levels of TMAO, its precursors (betaine, carnitine, deoxycarnitine, choline), and TMAO-to-precursor ratios are associated with clinical outcomes, including CVD and mortality. This was followed by an in-depth analysis of their genetic, gut microbial, and dietary determinants. The analyses were conducted in five Dutch prospective cohort studies including 7834 individuals. To further investigate association results, Mendelian Randomization (MR) was also explored. We found only plasma choline levels (hazard ratio [HR] 1.17, [95% CI 1.07; 1.28]) and not TMAO to be associated with CVD risk. Our association analyses uncovered 10 genome-wide significant loci, including novel genomic regions for betaine (6p21.1, 6q25.3), choline (2q34, 5q31.1), and deoxycarnitine (10q21.2, 11p14.2) comprising several metabolic gene associations, for example, CPS1 or PEMT. Furthermore, our analyses uncovered 68 gut microbiota associations, mainly related to TMAO-to-precursors ratios and the Ruminococcaceae family, and 16 associations of food groups and metabolites including fish-TMAO, meat-carnitine, and plant-based food-betaine associations. No significant association was identified by the MR approach. Our analyses provide novel insights into the TMAO pathway, its determinants, and pathophysiological impact on the general population.
Vaginal microbiota are associated with in vitro fertilization during female infertility
- 19 March 2024
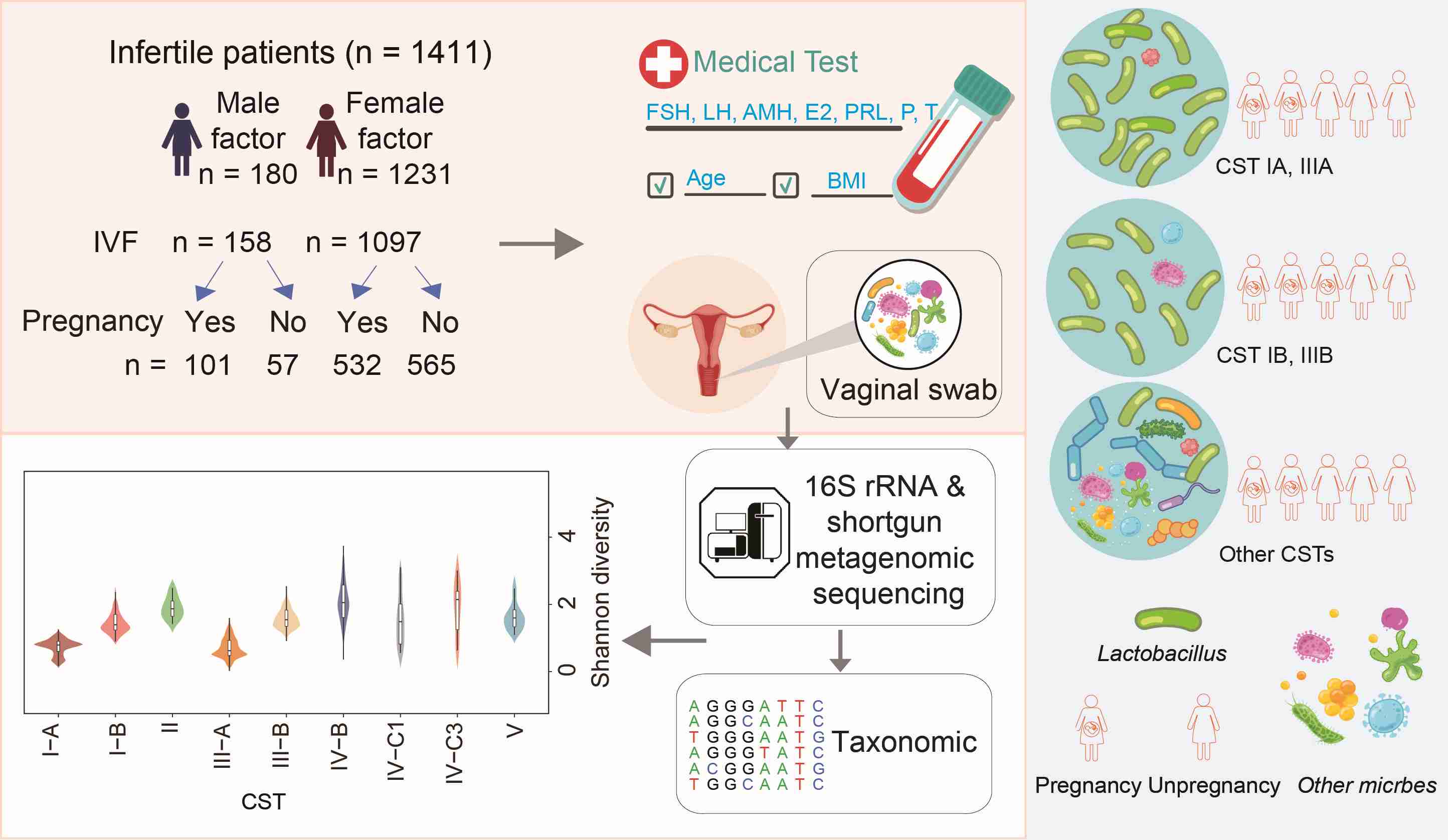
The vaginal microbiome is crucial for reproductive health, and as global infertility rates rise, understanding its impact on in vitro fertilization (IVF) is vital. Here, we analyzed the microbiome of 1411 individuals (1255 undergoing embryo transplantation) and revealed that a moderate abundance of Lactobacillus (~80%) is most beneficial for pregnancy (pregnancy rates were 54.35% and 57.73% for community state types [CST] I-B and III-B respectively), outperforming higher abundances (>90%) seen in I-A (44.81%) and III-A (51.06%). Metagenomic analysis of 71 samples indicates more antibiotic-resistance genes in nonpregnant women, with Proteobacteria and Firmicutes as predominant hosts. Variations among infertility groups suggest potential applications for detecting infertility and enhancing IVF outcomes using vaginal microbes. AMH, antimullerian hormones; BMI, body mass index; E2, estradiol; FSH, follicle-stimulating hormone; LH, luteinizing hormone; P, progesterone; PRL, prolactin; T, testosterone.
Arbuscular mycorrhizal fungal interactions bridge the support of root-associated microbiota for slope multifunctionality in an erosion-prone ecosystem
- 25 March 2024
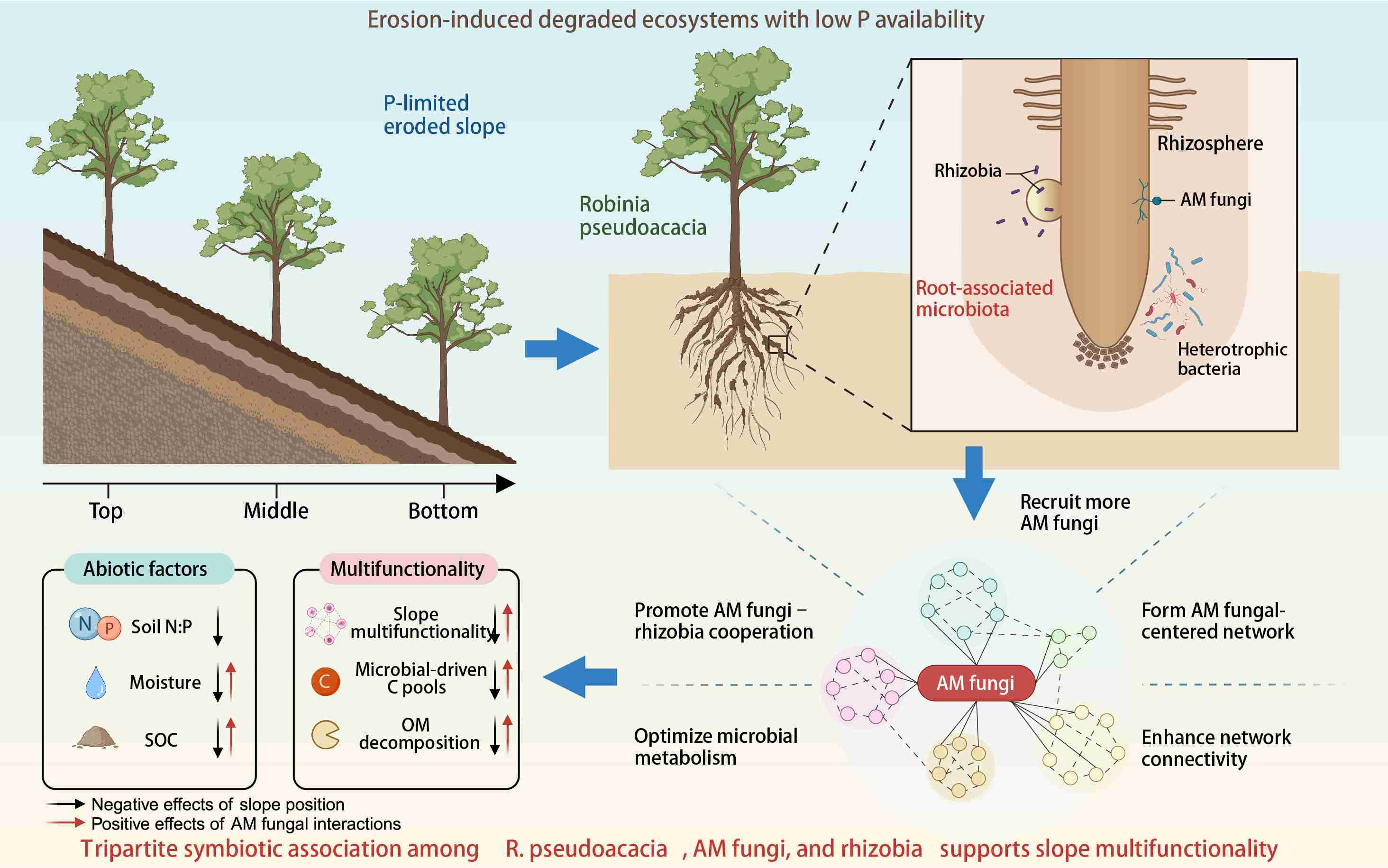
Robinia pseudoacacia strategically recruited arbuscular mycorrhizal (AM) fungi to cope with phosphorus limitation. AM fungi interacted with the assembly and composition of bacteria and rhizobia. AM fungal-centered underground network supported slope multifunctionality. The R. pseudoacacia–AM fungi–rhizobia association restores eroded ecosystems.
Clinical features and molecular landscape of cuproptosis signature-related molecular subtype in gastric cancer
- 05 April 2024
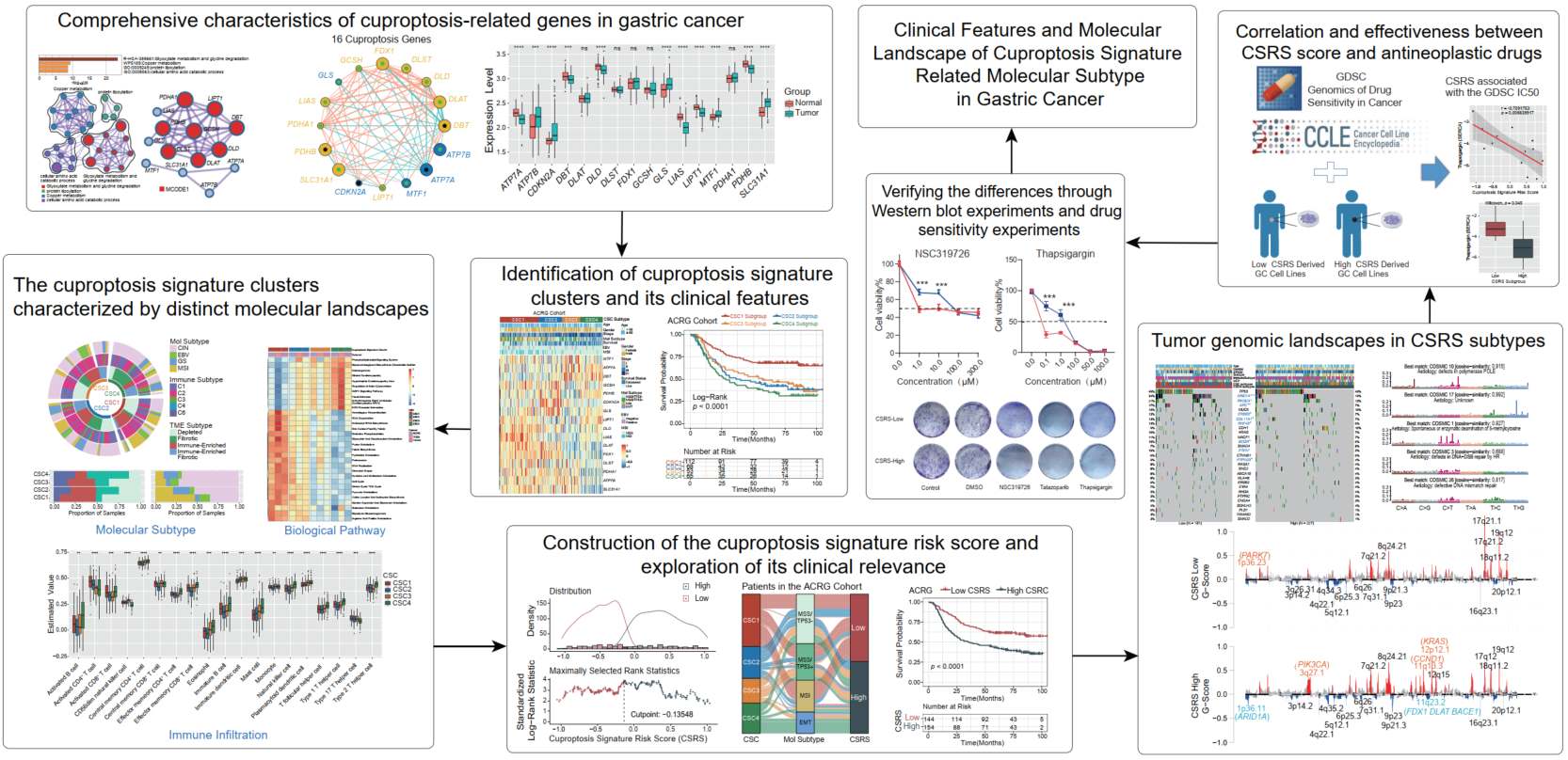
Four distinct cuproptosis signature-based clusters of gastric cancer (GC) were identified based on the expression pattern of curated cuproptosis genes, and associated with different clinical outcomes, biological pathways and highly consistent with distinct tumor immune contexture respectively. Based on the cuproptosis signature risk score (CSRS), GC patients with higher CSRS scores were characterized by decreased survival time and correlated with tumor adhesion state and lower tumor mutation loads. DBT, MTF1, or ATP7A were significantly elevated in the CSRS-High subtype, while ATP7B, SLC31A1, GCSH, LIAS, DLAT, FDX1, DLD, and PDHA1 were significantly enriched in the CSRS-Low subtype. Drug sensitivity analyses also revealed potential cancer compounds for GC with CSRS-High scores, and molecular biology experiments were validated.
A new evaluation system for drug–microbiota interactions
- 07 May 2024
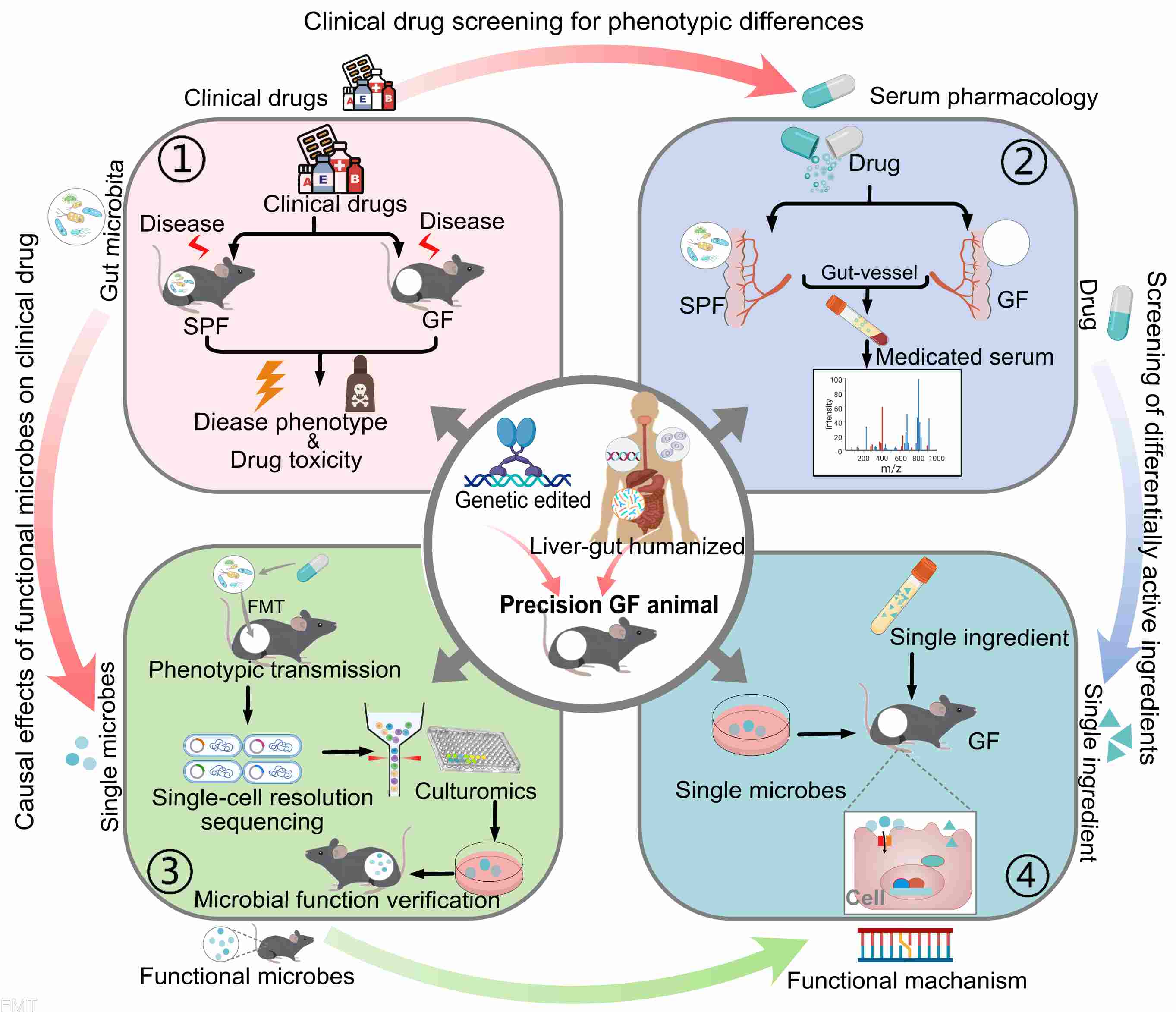
Here, we propose a new evaluation system and research strategy for drug microbial interaction. Leveraging precision germ-free animal models for the design of next-generation drug active/toxic ingredients involving gut microbiota, as well as microbes sorting and cultromics, to elucidate the exact mechanism of interaction between single microbe and single drug ingredient in vivo.
Cardiovascular disease therapeutics via engineered oral microbiota: Applications and perspective
- 09 May 2024
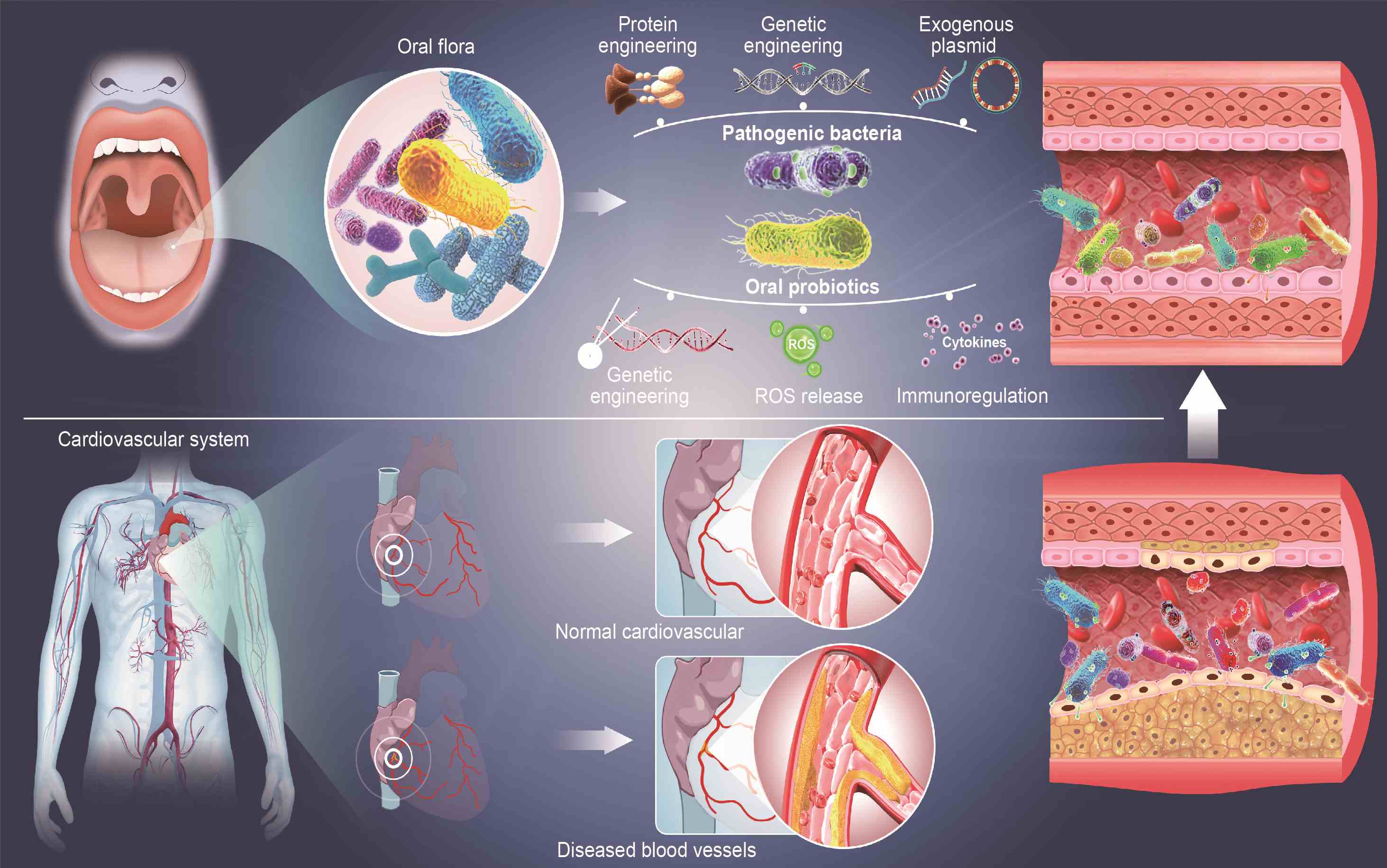
Engineering bacteria are considered as a potential treatment for cardiovascular diseases and related risk factors. Oral bacteria are closely related to the occurrence and development of cardiovascular diseases, and their engineering has broad prospects and potential in the treatment of cardiovascular diseases. Oral pathogenic bacteria undergo protein and genetic engineering, including the incorporation of exogenous plasmids to yield therapeutic effects; genetically engineered oral probiotics can be harnessed to secrete cytokines and reactive oxygen species, offering novel therapeutic avenues for cardiovascular diseases.
Understanding host immune responses in Clostridioides difficile infection: Implications for pathogenesis and immunotherapy
- 11 May 2024
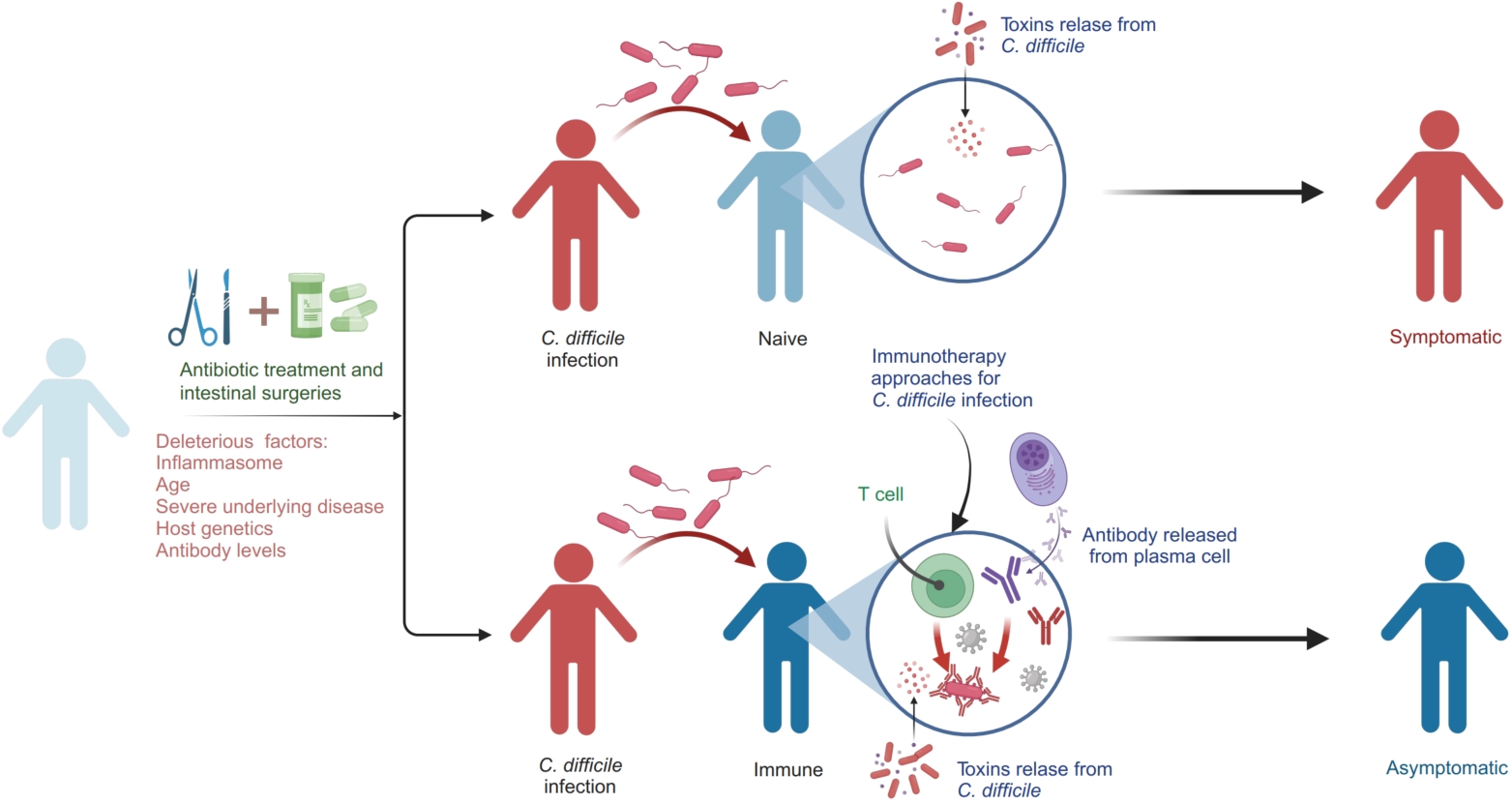
The progression of Clostridioides difficile infection (CDI) is shaped by multiple factors, such as antibiotic usage, age, comorbidities, immune gene polymorphisms, and antibody levels. Among these, the immune response stands out as a pivotal determinant in controlling both the course and severity of CDI. Leveraging immunotherapy presents substantial promise for advancing treatment and prevention strategies for CDI. This encompasses a range of approaches, including the use of monoclonal antibodies targeting C. difficile toxins, the development of vaccines to stimulate protective immune responses, and the application of fecal microbiota transplantation to restore microbial balance and bolster colonization resistance against C. difficile. These innovative immunotherapeutic interventions hold the potential to revolutionize CDI management by targeting key aspects of the disease's pathogenesis and immune regulation.
Long-term nitrogen input reduces soil bacterial network complexity by shifts in life history strategy in temperate grassland
- 15 April 2024
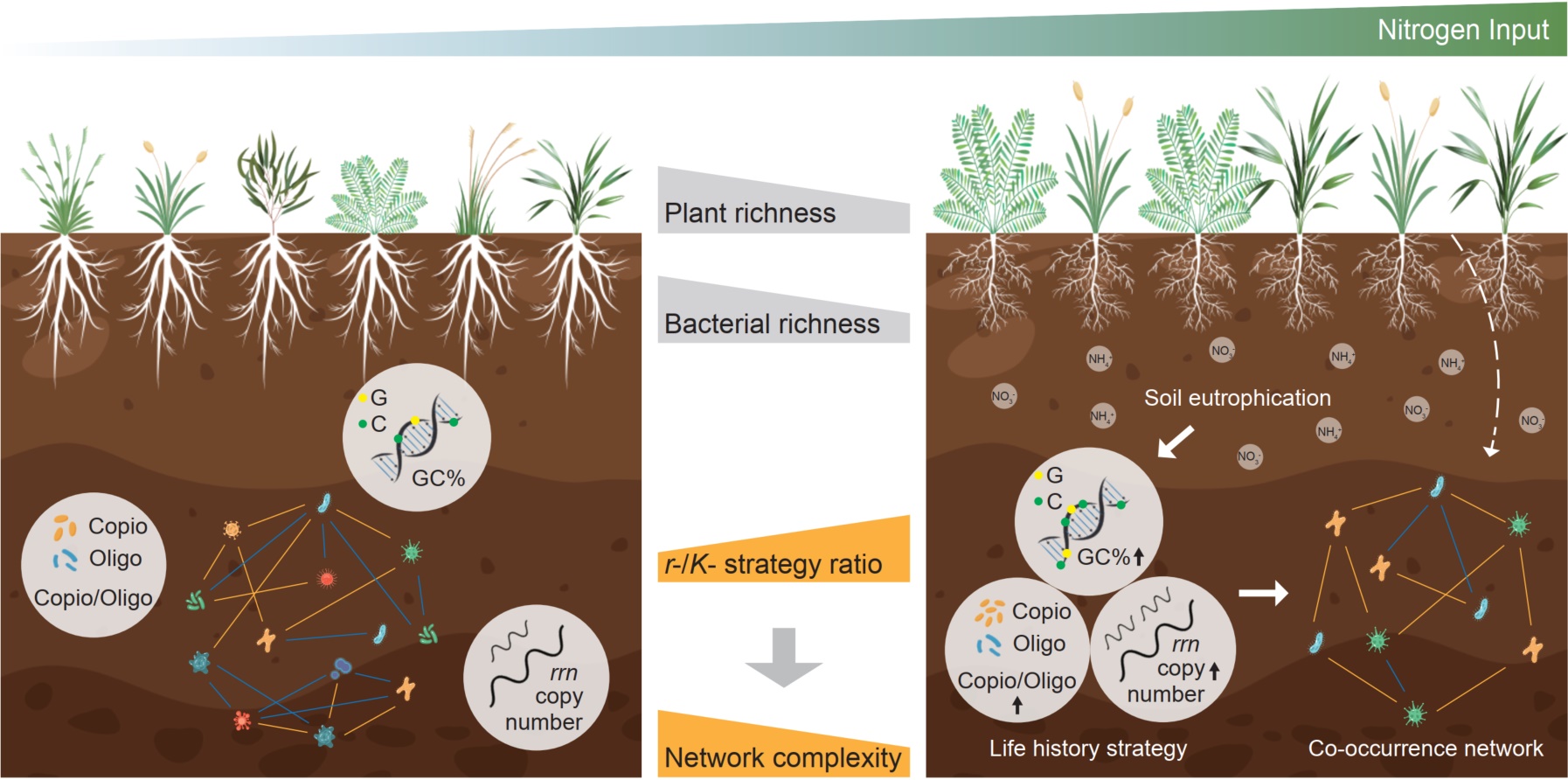
We investigated soil bacterial and fungal communities, constructed co-occurrence networks, and estimated bacterial traits along a gradient of nitrogen (N) input. The results showed that soil bacterial co-occurrence networks complexity decreased with increasing N input. The ratio of negative to positive cohesion decreased with increasing N input, suggesting the declined competitive but strengthened cooperative interactions. However, soil fungal network complexity did not change under N enrichment. In addition, N input stimulated the copiotroph/oligotroph ratio, ribosomal RNA operon (rrn) copy number, and guanine-cytosine (GC) content of soil bacteria, shifting bacterial life history strategy toward copiotroph with increased r-/K-strategy ratio. Piecewise structural equation modeling results further revealed that the reduction in bacterial co-occurrence network complexity was directly regulated by the increased bacterial r-/K-strategy ratio, rather than reduced bacterial richness. Our study reveals the mechanisms through which microbial traits regulate interactions and shape co-occurrence networks under global changes.
Robust CRISPR/Mb2Cas12a genome editing tools in cotton plants
- 04 June 2024
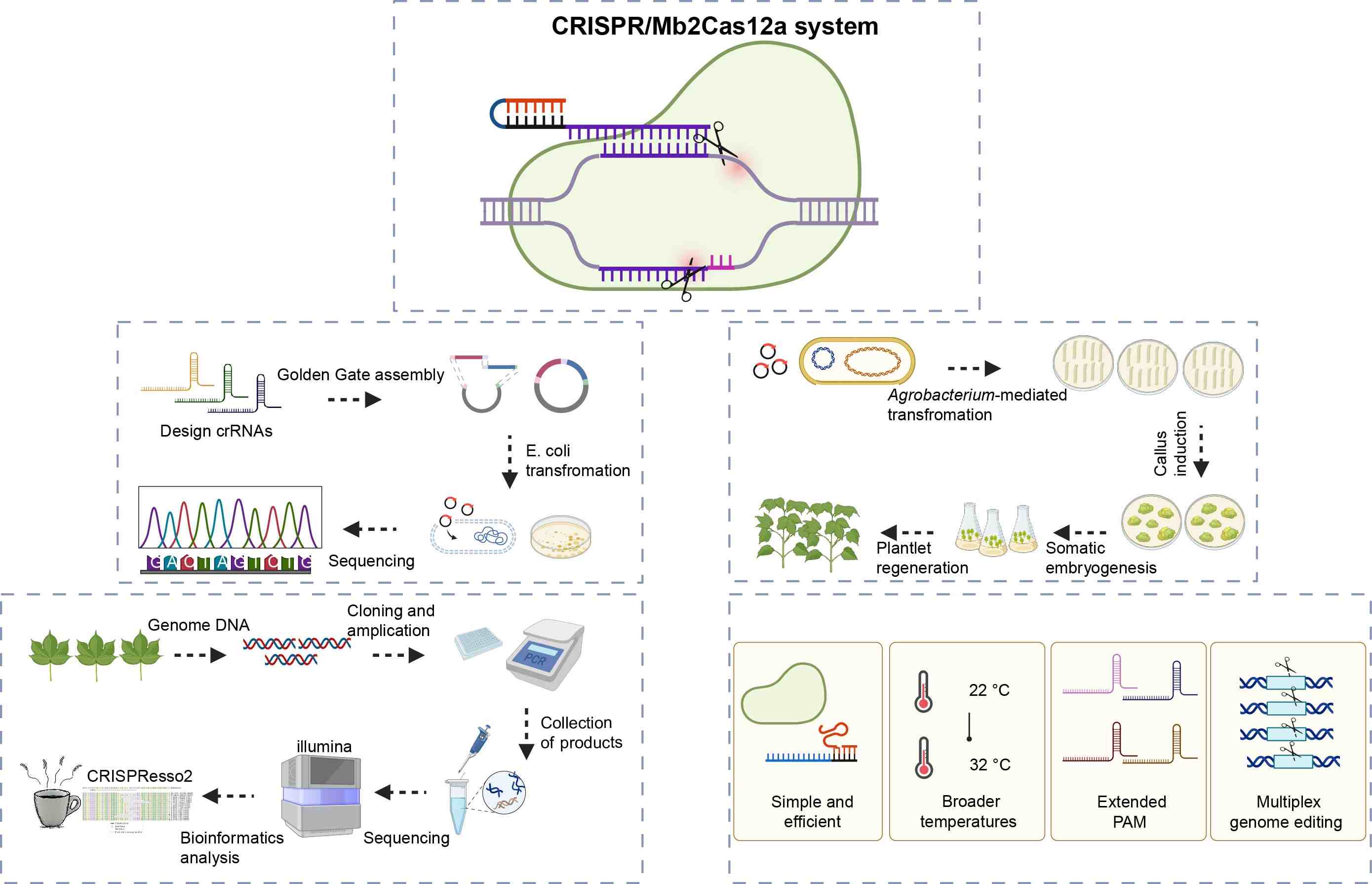
The efficiency and accuracy of the CRISPR/Mb2Cas12a system were demonstrated in cotton, achieving an efficiency of over 90% at target sites. Notably, Mb2Cas12a exhibited significant tolerance under different temperatures ranging from 22°C to 32°C. Additionally, the Mb2Cas12a system revealed effective editing at more relaxed VTTV PAM sites in the cotton genome, which expanded the genome editing range by approximately 2.6-fold than the wide-type LbCas12a. Finally, a multiplex genome editing system was also developed based on Mb2Cas12a, enabling simultaneous editing of eight target sites using a single crRNA cassette.
MASH-Ocean 1.0: Interactive platform for investigating microbial diversity, function, and biogeography with marine metagenomic data
- 19 May 2024
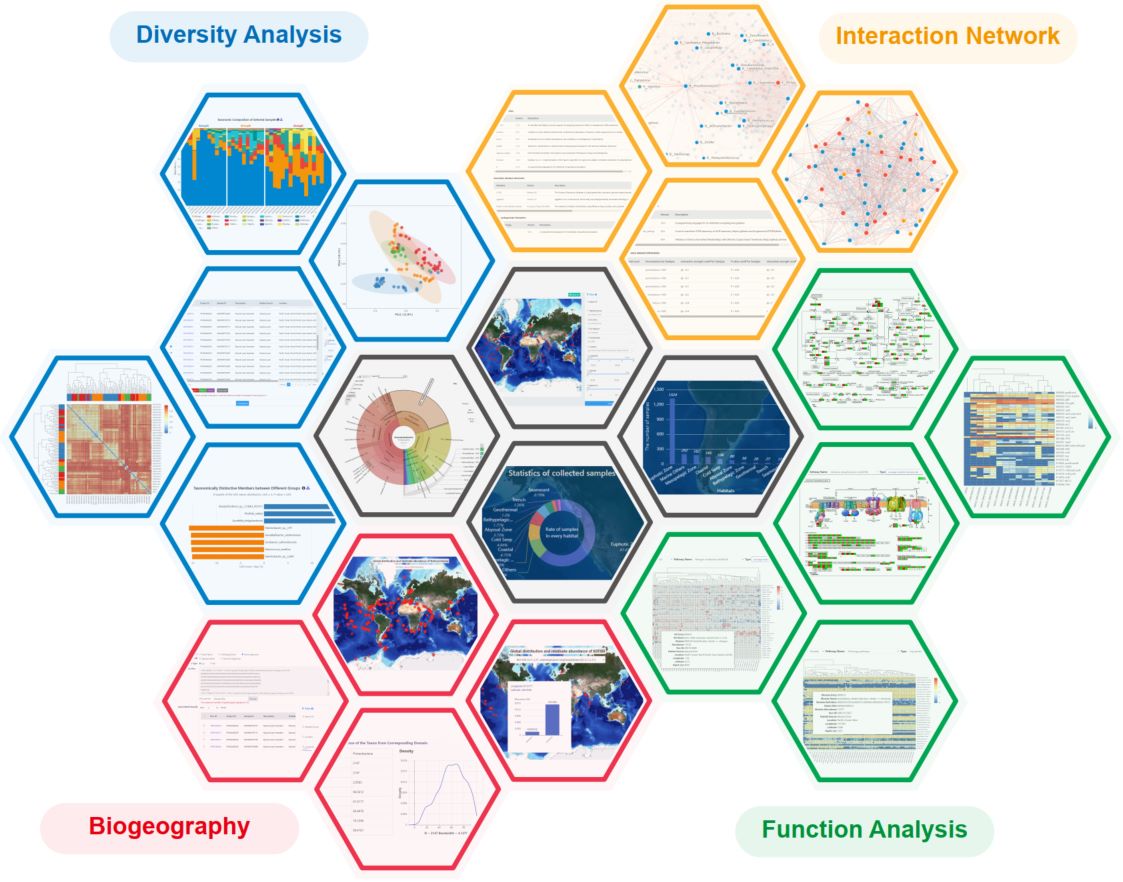
A large number of oceanic metagenomic data and environmental metadata have been published. However, most studies focused on limited ecosystems using different analysis tools, making it challenging to integrate these data into robust results and comprehensive global understanding of marine microbiome. Here, we constructed a systematic and quantitative analysis platform, the Microbiome Atlas/Sino-Hydrosphere for Ocean Ecosystem (MASH-Ocean: https://www.biosino.org/mash-ocean/), by integrating global marine metagenomic data and a unified data processing flow. MASH-Ocean 1.0 comprises 2147 metagenomic samples with five analysis modules: sample view, diversity, function, biogeography, and interaction network. This platform provides convenient and stable support for researchers in microbiology, environmental science, and biogeochemistry, to ensure the integration of omics data generated from hydrosphere ecosystems, to bridge the gap between elusive omics data and biological, ecological, and geological discovery, ultimately to foster the formation of a comprehensive atlas for aquatic environments.
GutUDB: A comprehensive multiomics database for intestinal diseases
- 27 April 2024
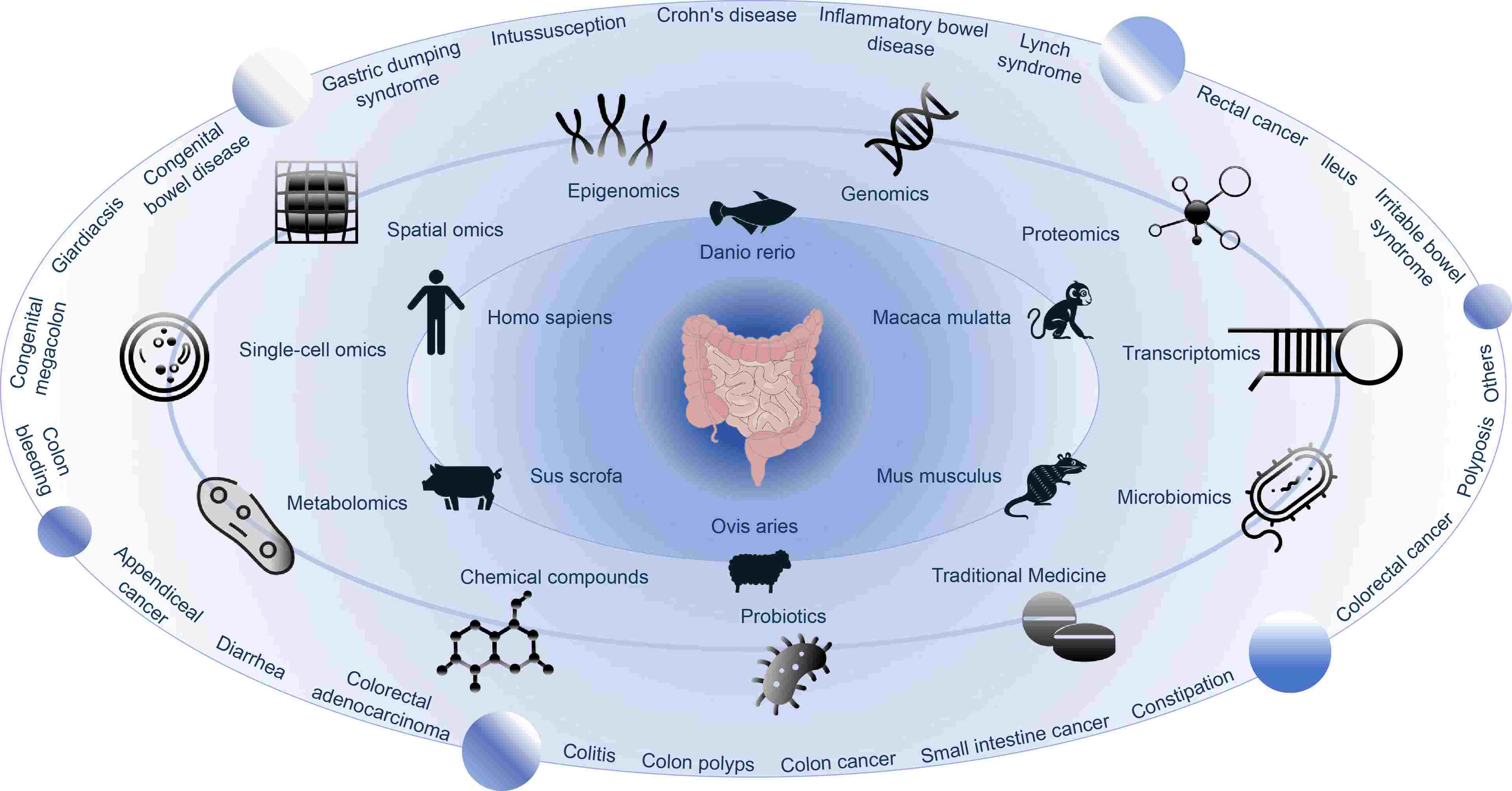
Gut Universe Database (GutUDB) provides a comprehensive, systematic, and practical platform for researchers, and is dedicated to the management, analysis, and visualization of knowledge related to intestinal diseases. Based on this database, eight major categories of omics data analyses are carried out to explore the genotype-phenotype characteristics of a certain intestinal disease. The first tool for comprehensive omics data research on intestinal diseases will help each researcher better understand intestinal diseases.
Intestinal linoleic acid contributes to the protective effects of Akkermansia muciniphila against Listeria monocytogenes infection in mice
- 27 April 2024
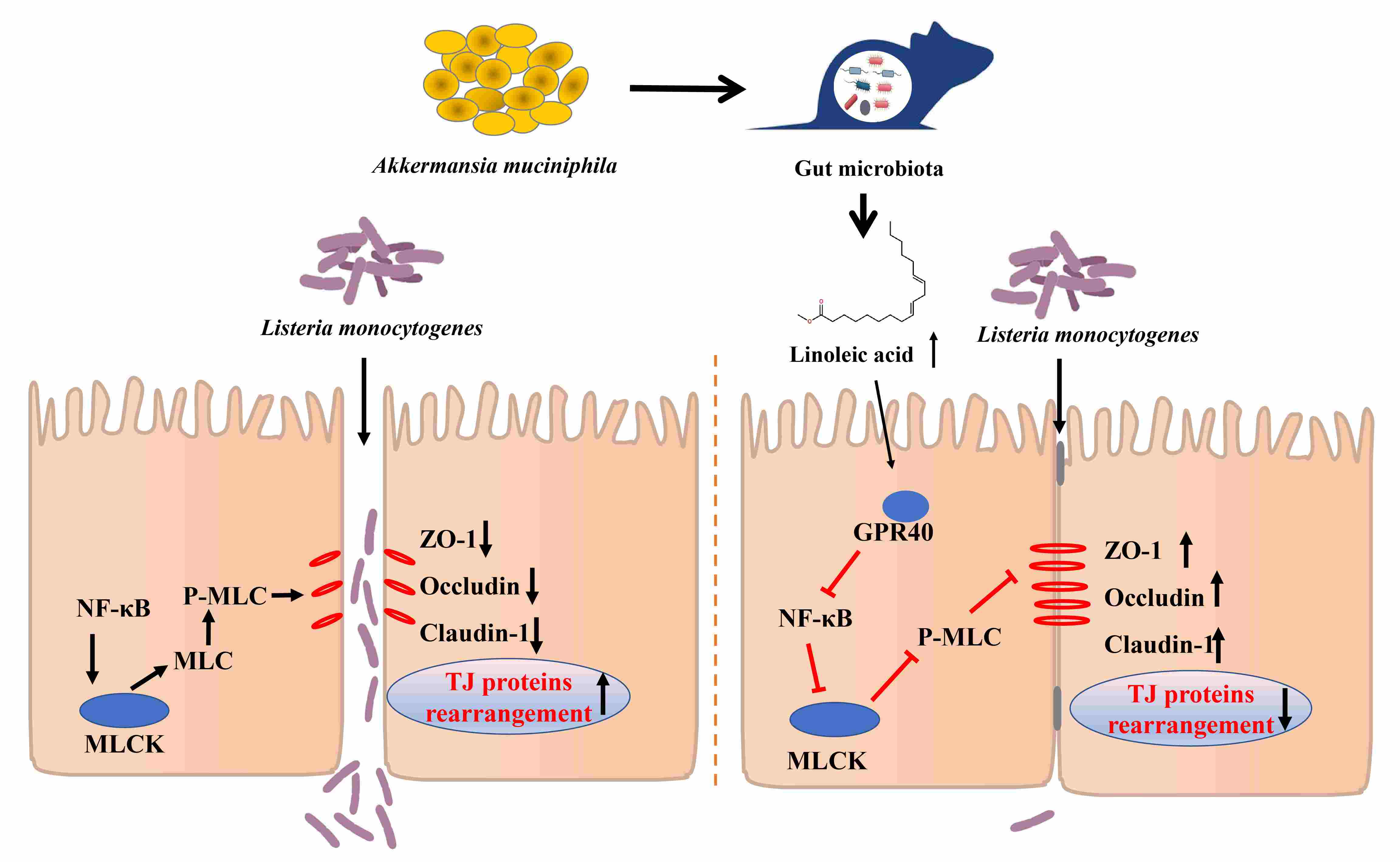
Akkermansia muciniphila pretreatment mitigated Listeria monocytogenes infection in mice. A. muciniphila improved gut microbiota disturbed by L. monocytogenes infection and significantly increased the level of intestinal linoleic acid in mice. Linoleic acid strengthened the intestinal epithelial barrier and reduced pathogen translocation partly by regulating NF-κB/MLCK pathway in a GPR40-dependent manner.
The feasibility of using pathobiome strains as live biotherapeutic products for human use
- 26 May 2024
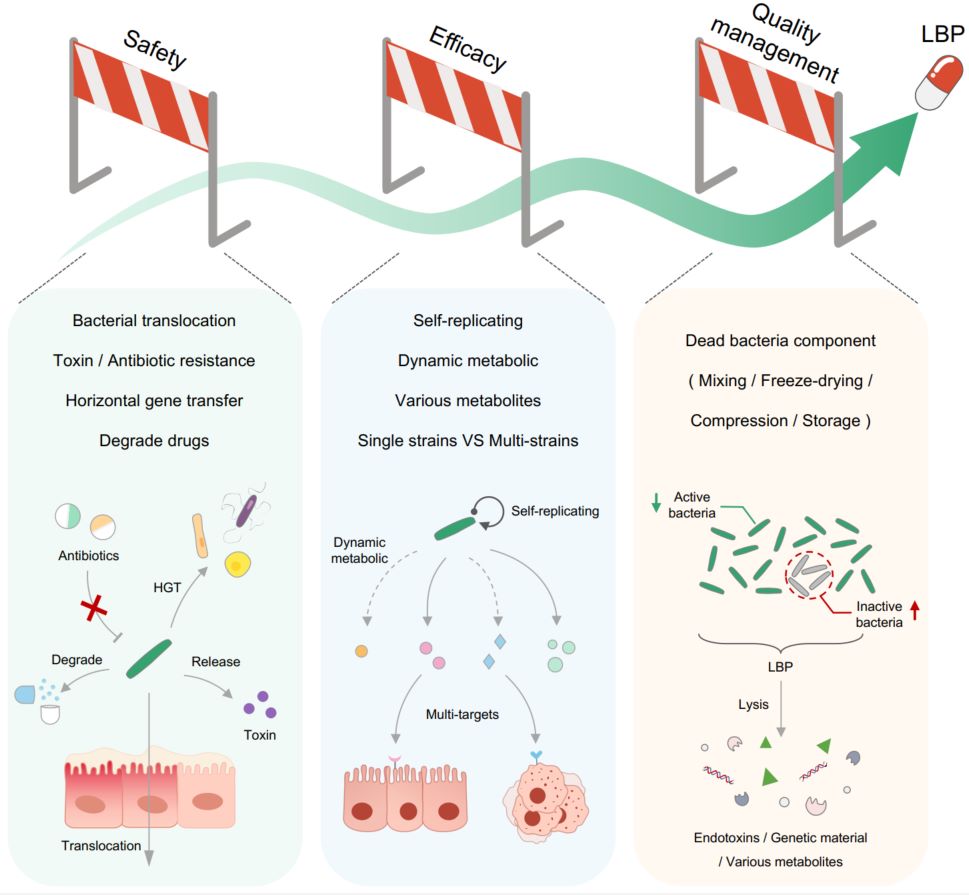
The evaluation of pathobiome strains should be conducted at the strain level, involving the identification of the functional genes, while considering the impact of ecological niche and drug interactions. The safety, efficacy, and quality management of live biotherapeutic products (LBPs), especially pathobiome strains, have certain peculiarities. Promising development methods include the recombinant LBP and active metabolites.












