











Strategies and mechanisms of precision genome engineering: From gene editing to genome writing
- 26 July 2026
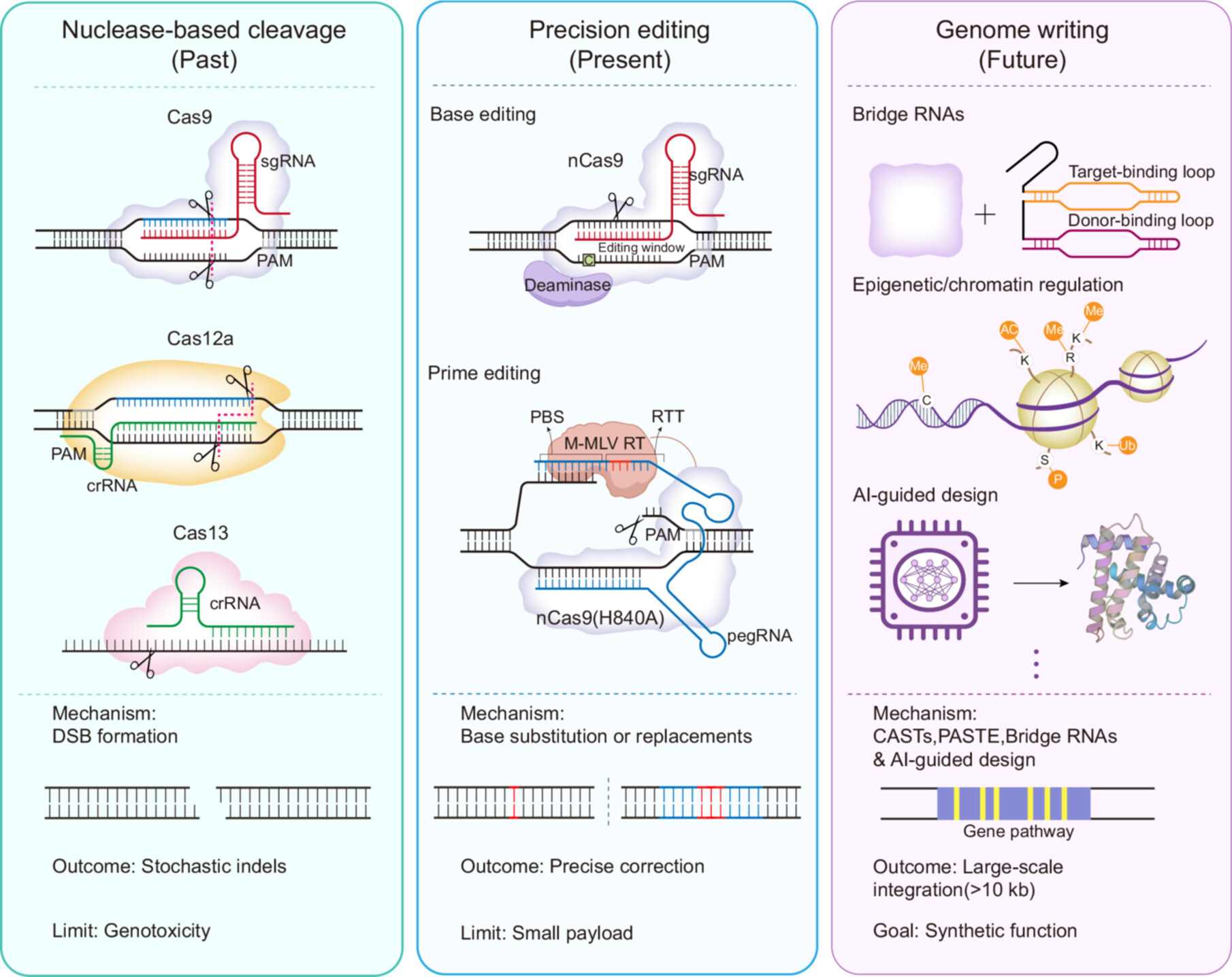
In this review, we examined the progression of genome manipulation from stochastic nuclease-mediated cutting toward precise editing and programmable genome writing. We discussed tools like multi-kilobase RNA-guided integrators and Artificial Intelligence (AI)-designed effectors and showed how these advances enable researchers to treat genomes as programmable media for synthetic design and Generative Biology.
Bile acid metabolism and signaling in human and animal health: A One Health perspective
- 17 July 2026
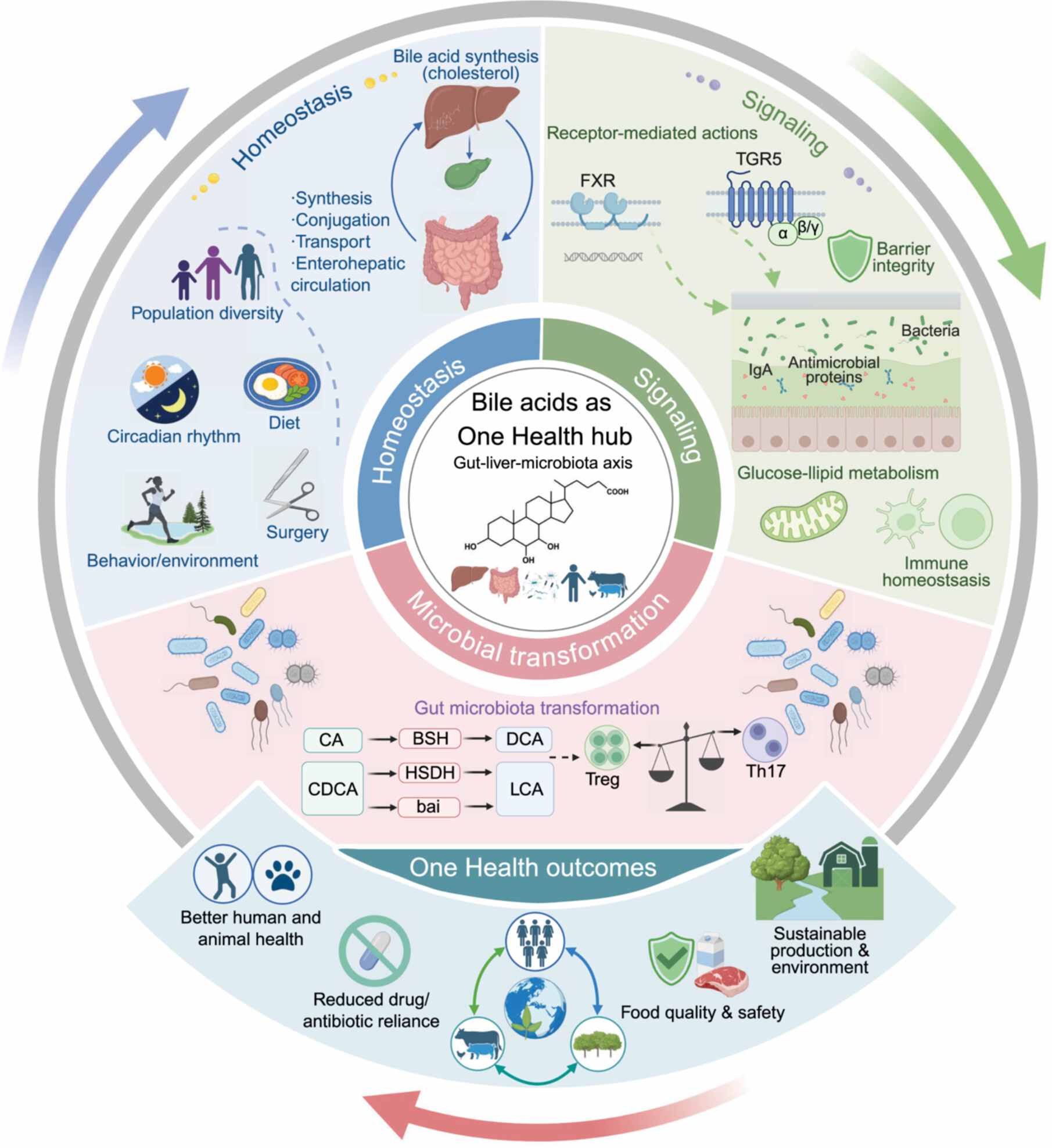
Bile acids maintain host homeostasis through enterohepatic circulation, microbial transformation, and bile acid receptor signaling, thereby regulating glucose and lipid metabolism, intestinal barrier integrity, and immunity. These effects support human disease intervention and health-oriented animal production, reducing drug dependence and residue risks while promoting food safety, sustainability, and One Health benefits.
Sexual dimorphism in hormone–gut microbiome associations across reproductive states in wild giant pandas
- 05 June 2026
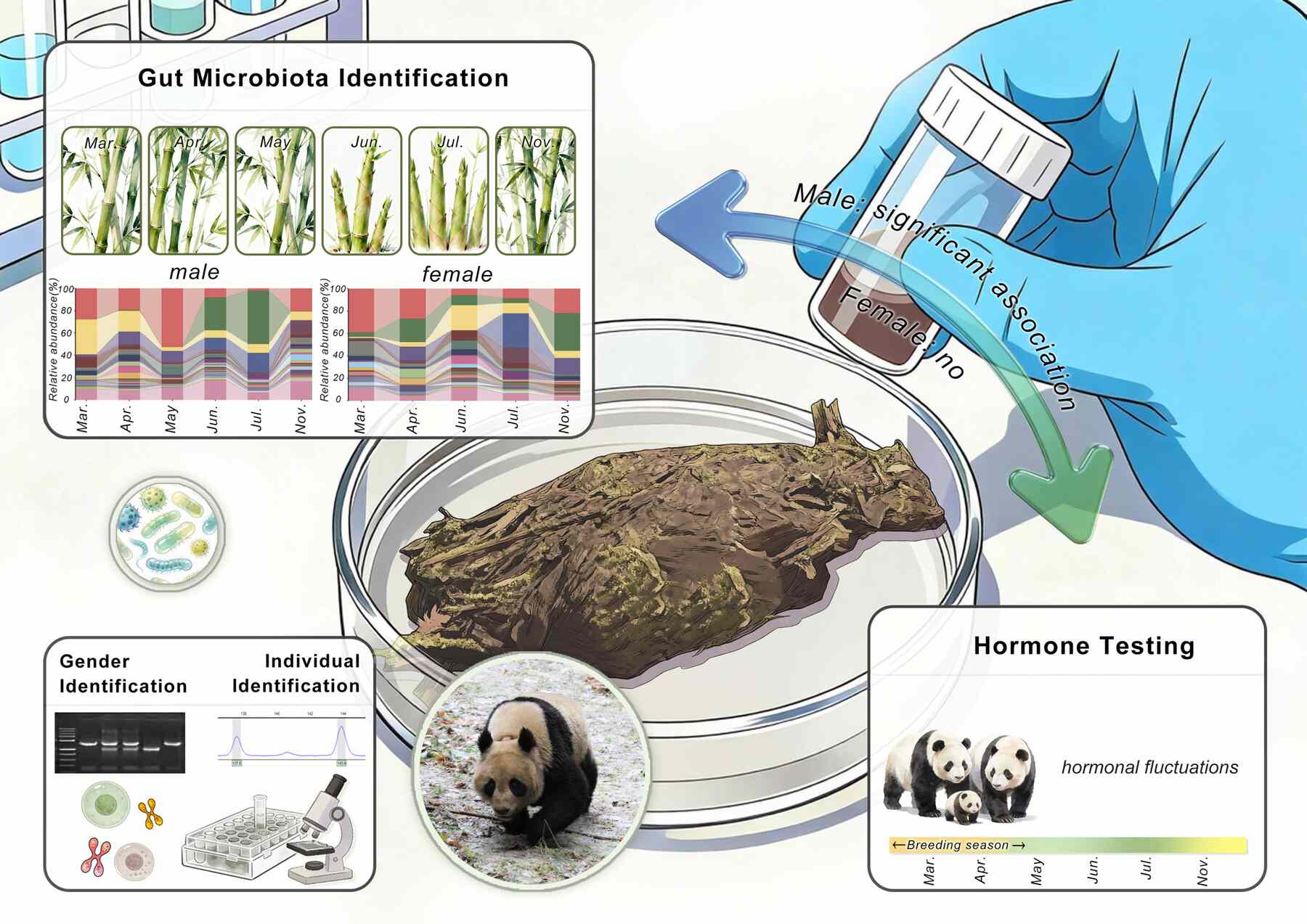
Gut microbiota–host endocrine interactions in wild mammals remain poorly understood. Giant pandas experience seasonal dietary shifts and constrained annual reproduction, offering an ideal model to investigate microbiome–endocrine coupling. We integrated fecal hormone profiling, microsatellite-based individual identification, and gut microbiome sequencing in wild giant pandas. Microbial diversity and composition varied seasonally, with α diversity peaking during bamboo shoot-feeding. Hormonal dynamics followed predictable patterns: elevated estrogen during estrus, sustained progesterone post-estrus, and higher cortisol during leaf/stem-feeding periods. Hormone levels significantly correlated with microbial taxa and diversity, indicating coordinated microbiome–endocrine dynamics. Notably, microbiome–hormone coupling was significant in males but not in females, and males exhibited greater hormonal and microbial variability. Our findings demonstrate seasonal restructuring of gut microbiome and endocrine profiles in wild giant pandas with sex-dependent coupling, providing an integrated framework for non-invasive conservation monitoring.
Microbial ecological memory of recurrent drought enhances host stress tolerance through enrichment of plant-beneficial bacteria
- 15 June 2026
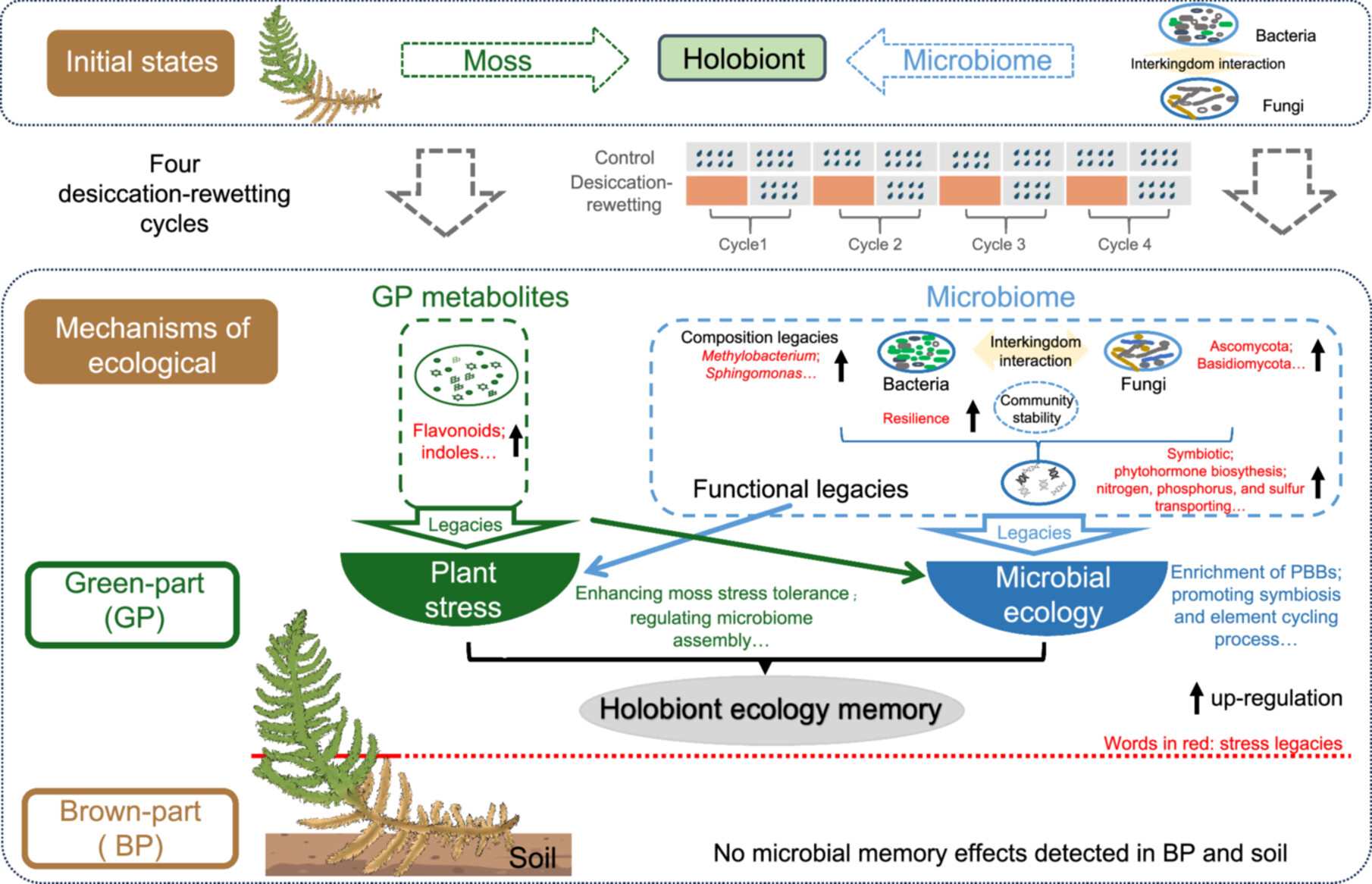
Recurrent desiccation induced niche-specific microbial ecological memory in the green part of Calohypnum plumiforme, marked by enrichment of plant-beneficial bacteria, especially Methylobacterium and Sphingomonas, and functional shifts toward symbiosis and stress protection, potentially enhancing host tolerance within a “holobiont ecology memory” framework.
RNA viral protein structure database: A comprehensive and user-friendly web database for RNA viral protein structures
- 13 July 2026
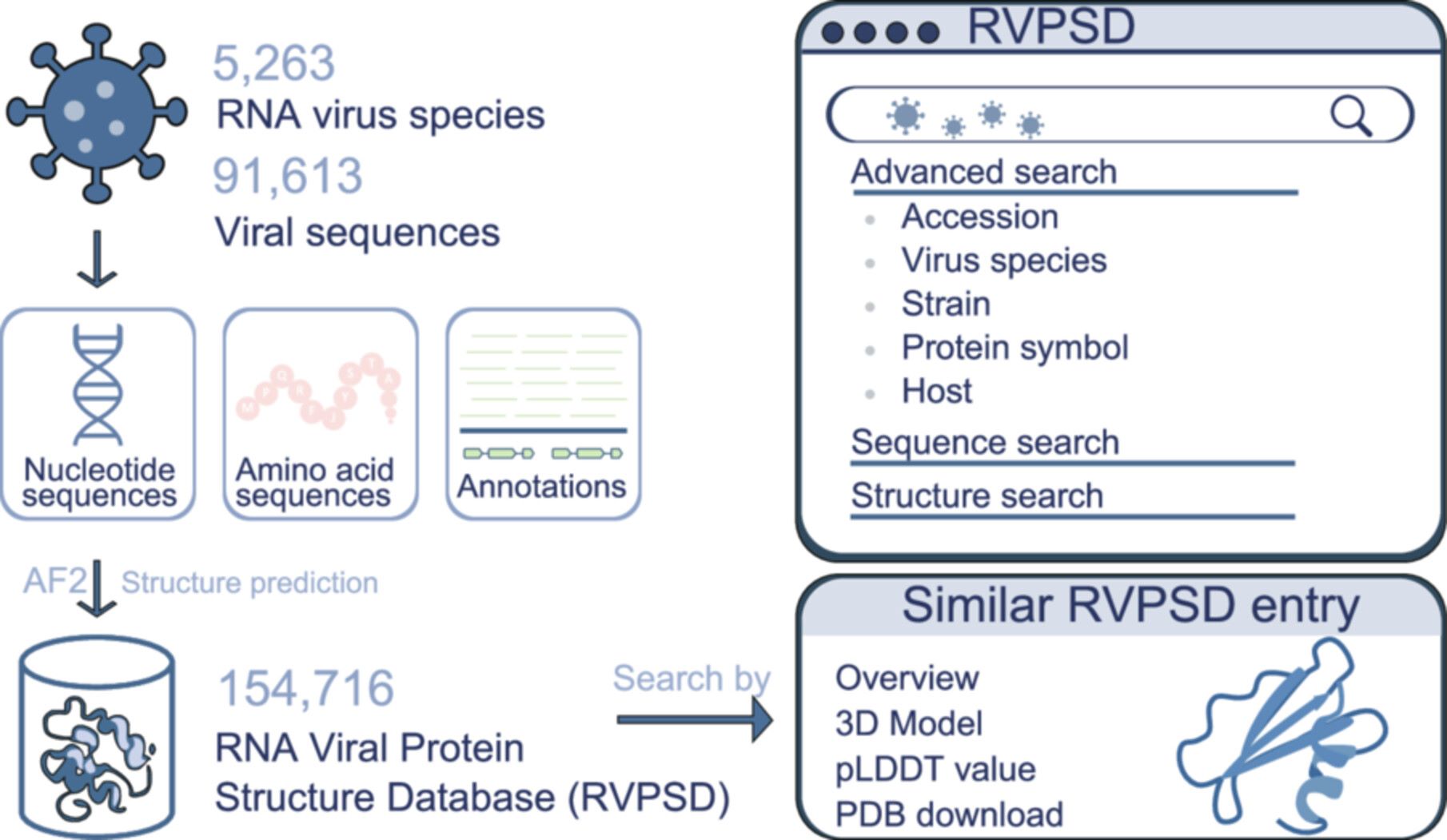
Given the central role of protein structures in RNA virus biology, the RNA viral protein structure database (RVPSD, https://virus.9itsg.net) integrates taxonomic classification, viral nucleotide and protein sequences, functional annotations, and predicted three-dimensional protein structures. Currently, RVPSD comprises 154,716 AlphaFold2-predicted RNA viral protein structures from 5263 RNA virus species, with > 80% exhibiting high predicted local distance difference test scores. RVPSD also provides a searchable and interactive web interface to support comparative structural analyses and functional annotation of viral dark matter.
Warm-wet transition restructures arid lake microbiomes and functional potential
- 08 July 2026
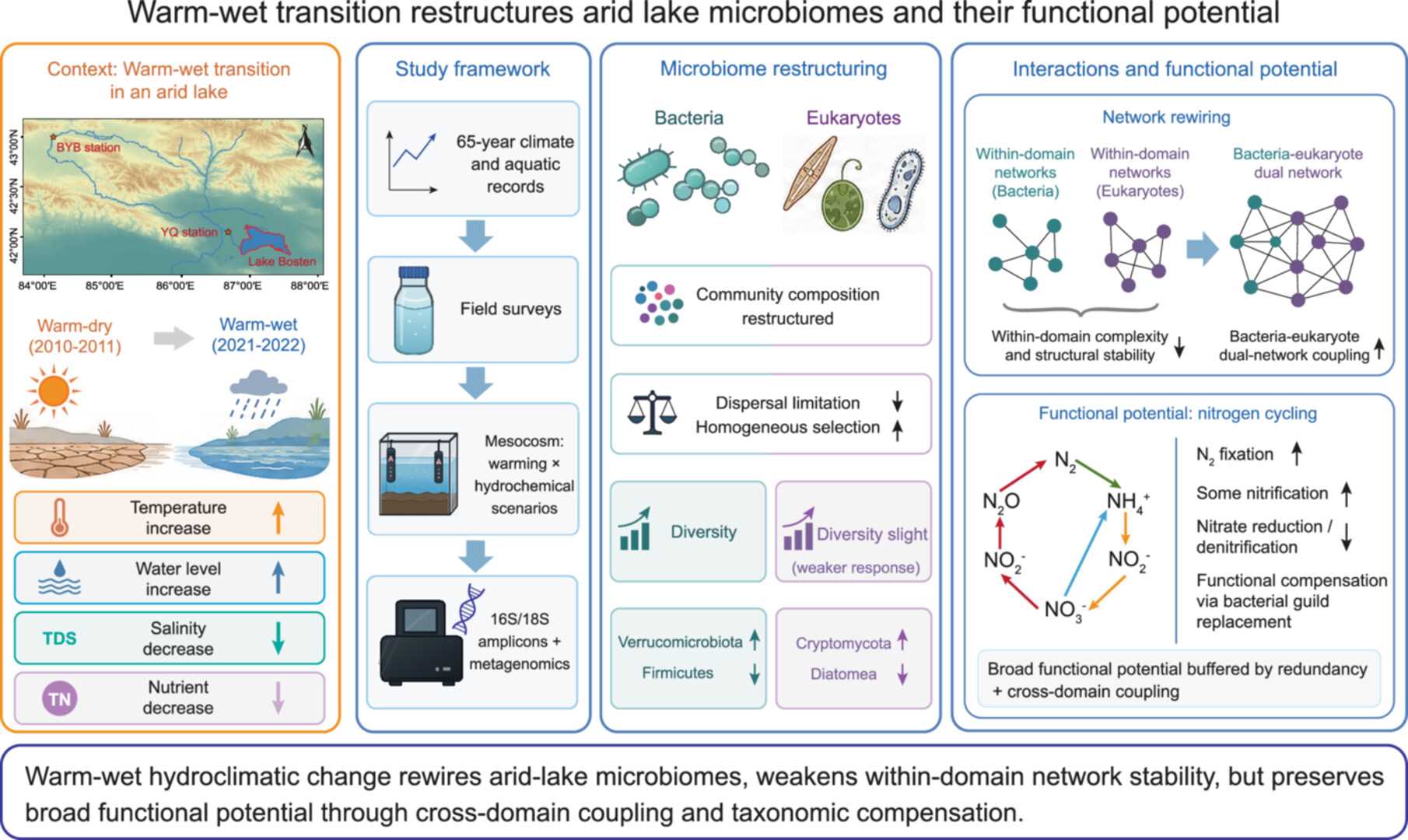
This study summarizes how a long-term warm-dry to warm-wet transition restructures microbiomes and functional potential in Lake Bosten, an arid lake in northwestern China. Warm-wet hydroclimatic change increased water level and hydrological connectivity, while decreasing salinity and total nitrogen. These environmental shifts restructured both bacterial and eukaryotic communities, with reduced dispersal limitation and enhanced homogeneous selection. Bacterial diversity increased, whereas eukaryotic diversity showed a weaker response. Warm-wet conditions weakened within domain community compositional stability and network structural stability, but strengthened bacteria-eukaryote interactions. Despite pronounced taxonomic and network reorganization, broad functional potential was buffered through functional redundancy, cross domain coupling, and taxonomic replacement within bacterial functional guilds, particularly in nitrogen cycling pathways.
Harnessing microbial metabolism and nutrition to target the gut–mammary axis for better livestock production and health
- 09 July 2026
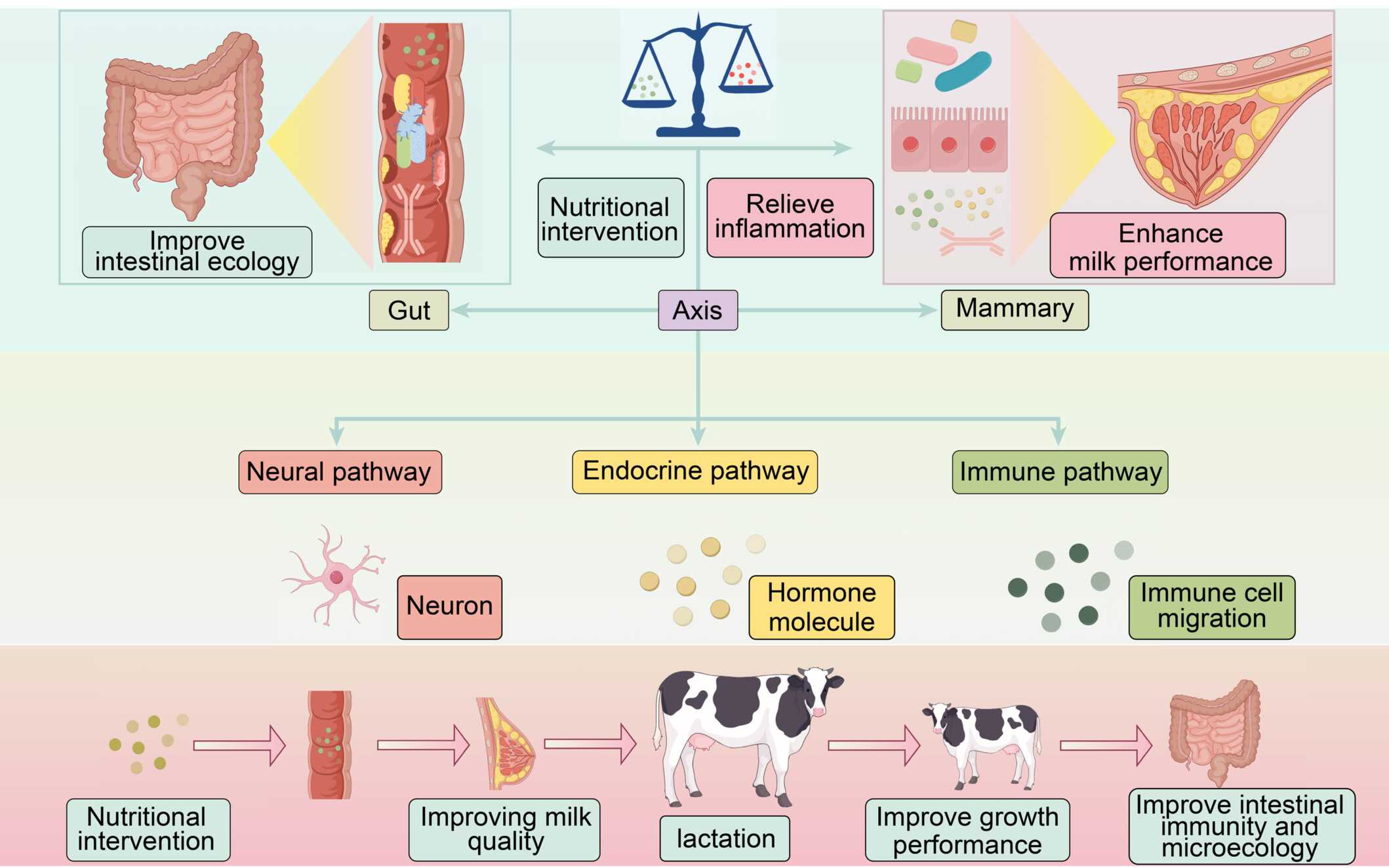
The gut–mammary axis integrates intestinal health with mammary function through neural, endocrine, and immune pathways. Nutritional interventions (probiotics, prebiotics, metabolites) improve gut microecology, generating microbial signals that travel via the axis to the mammary gland. There, they relieve inflammation, enhance milk quality and lactation, and boost offspring growth. This framework highlights precision nutrition as a strategy to modulate the gut–mammary axis for sustainable livestock production.
A synthetic community promotes macadamia growth via root-microbe interactions and systemic plant responses
- 09 July 2026
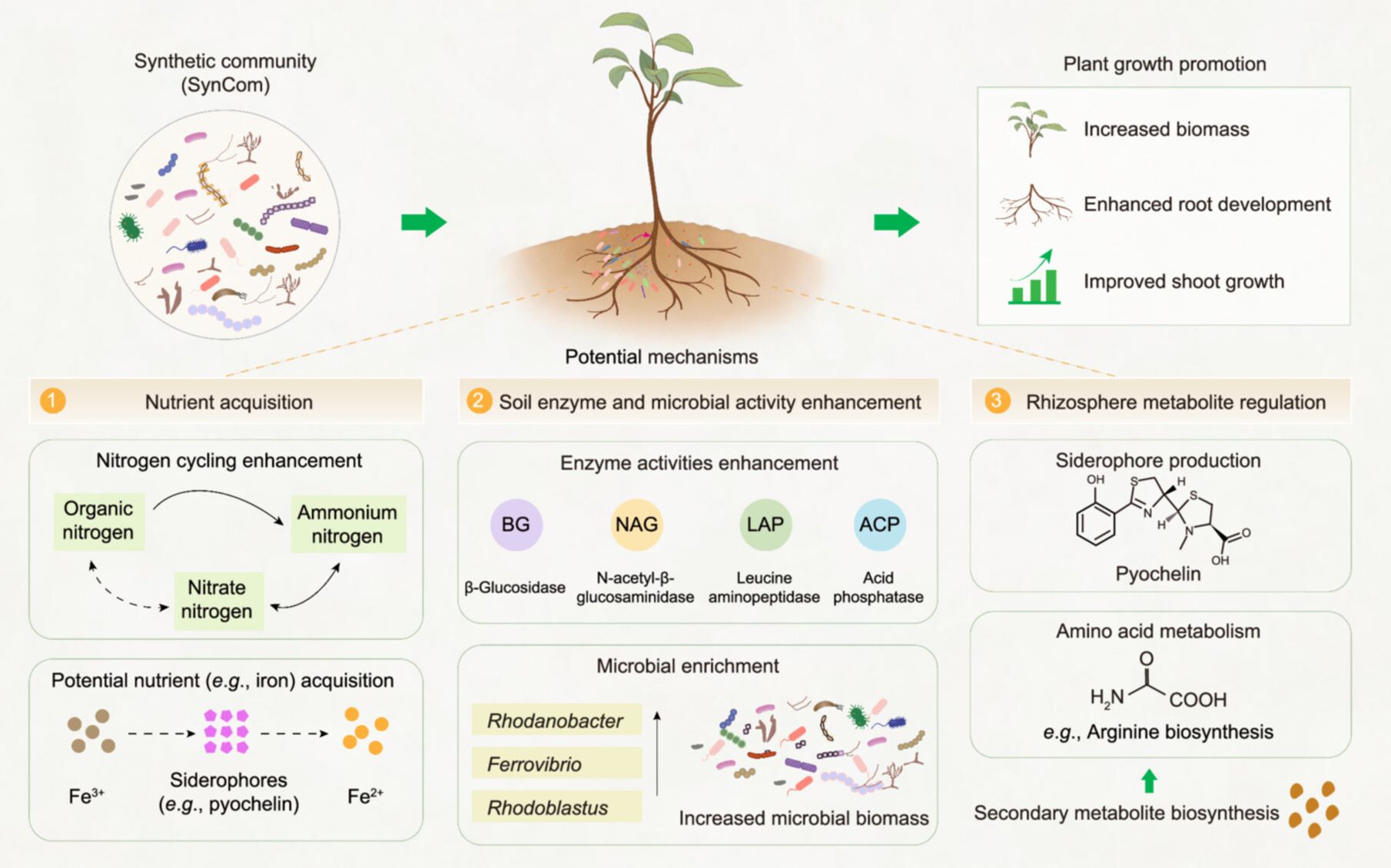
We employed an 18-month pot experiment and an integrated multi-omics approach to elucidate the potential mechanisms underlying synthetic community (SynCom)-mediated macadamia growth promotion. SynCom inoculation enhanced soil nitrogen cycling and potential nutrient (e.g., iron) acquisition. It simultaneously increased soil extracellular enzyme activities and enriched beneficial bacteria such as Rhodanobacter, Ferrovibrio, and Rhodoblastus in the root compartment. In addition, SynCom inoculation promoted the accumulation of rhizosphere metabolites, including the microbial siderophore pyochelin, and drove the co-enrichment of transcriptomic and metabolomic pathways, notably the arginine biosynthesis pathway. This coordinated response may have stimulated nitrogen and iron cycling in the soil, thereby promoting the healthy growth of macadamia.
Microbiome-mediated plant-soil feedbacks as a tool for resilient agriculture
- 03 July 2026
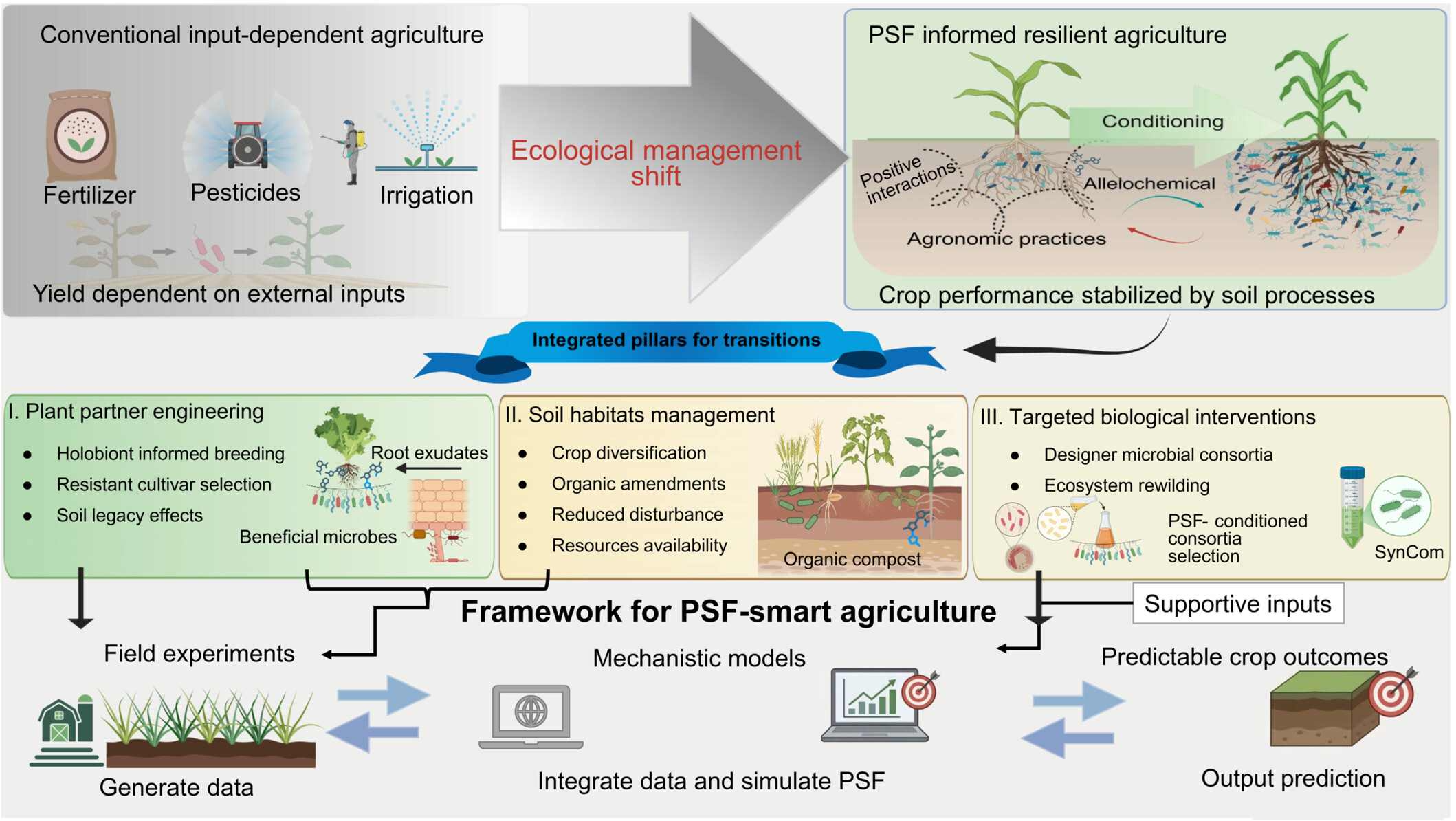
Translating plant-soil feedbacks (PSFs) into reliable agricultural management requires shifting from input-dependent practices toward endogenous, process-driven crop resilience. To bridge this gap, we propose a predictive framework centered on three strategic interventions: engineering the crop holobiont, diversifying the soil habitat, and deploying targeted microbial consortia. Rather than relying on static correlations, this framework integrates field-scale gradients with mechanistic models to validate soil legacies dynamically. By closing this empirical-predictive loop, we can turn complex ecological principles into quantifiable, actionable designs for climate-resilient cropping systems.
A multiplex interactome of Ebola virus proteins reveals TM9SF2 as a cell-surface attachment factor that promotes viral entry
- 02 July 2026
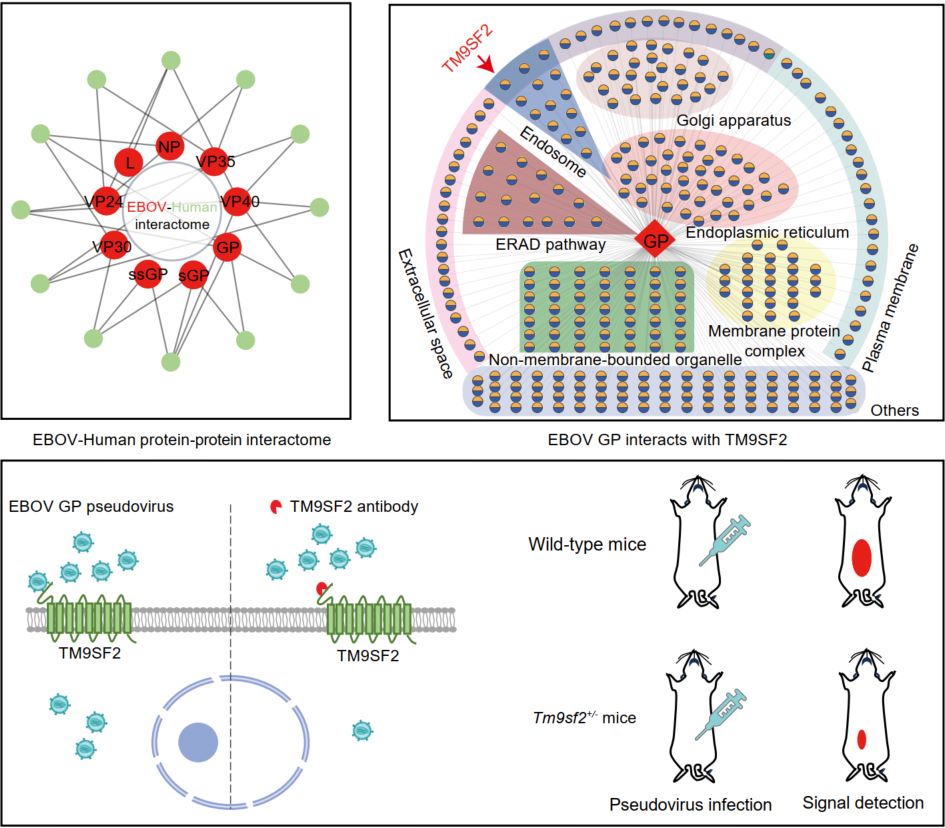
This study generates a comprehensive Ebola virus (EBOV)-human protein–protein interactome, comprising 1728 core high-confidence interactions. Further interactome analysis revealed the potential association of EBOV glycoprotein (GP) with the host factor TM9SF2. Subsequent mechanistic investigations confirmed that TM9SF2 functions as an attachment factor that, through its association with EBOV GP, promotes viral attachment and internalization to facilitate infection. These findings position TM9SF2 as a promising antiviral target.
Host-specific and deterministic microbiome assembly in major coleopteran stored-grain pests
- 11 June 2026
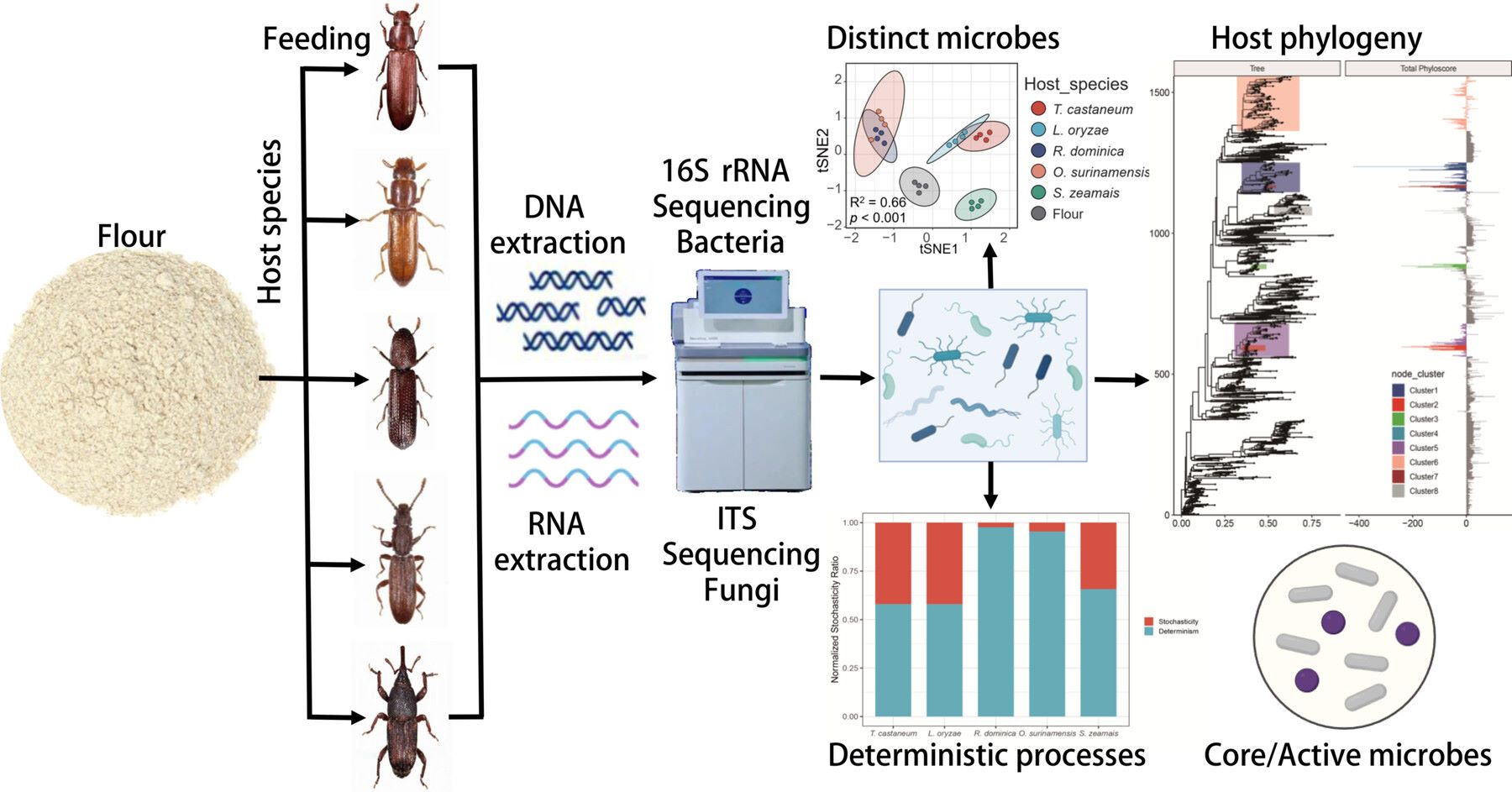
Integrating multiplexed immunofluorescence, animal models, and clinical samples, our single-cell and spatial atlas maps tumor microenvironment evolution during bladder cancer (BCa) progression. We reveal that stemness-associated tumor cells (SDC1+), POSTN+ myofibroblastic cancer-associated fibroblasts (mCAFs), and immunosuppressive monocytic myeloid-derived suppressor cells (M-MDSCs) are enriched in high-grade BCa. Mechanistically, mCAF-derived FN1 and M-MDSC-derived NAMPT engage endothelial integrin α5β1 to drive angiogenesis. Subsequently, endothelial cells sustain tumor stemness via the COL4A1/2–SDC1 axis. Therapeutically, targeting integrin α5β1 synergizes with immune checkpoint blockade, highlighting a promising stroma-directed strategy.
Integrin α5β1-mediated multicellular crosstalk in the tumor microenvironment drives bladder cancer progression and reveals targetable vulnerabilities
- 23 June 2026
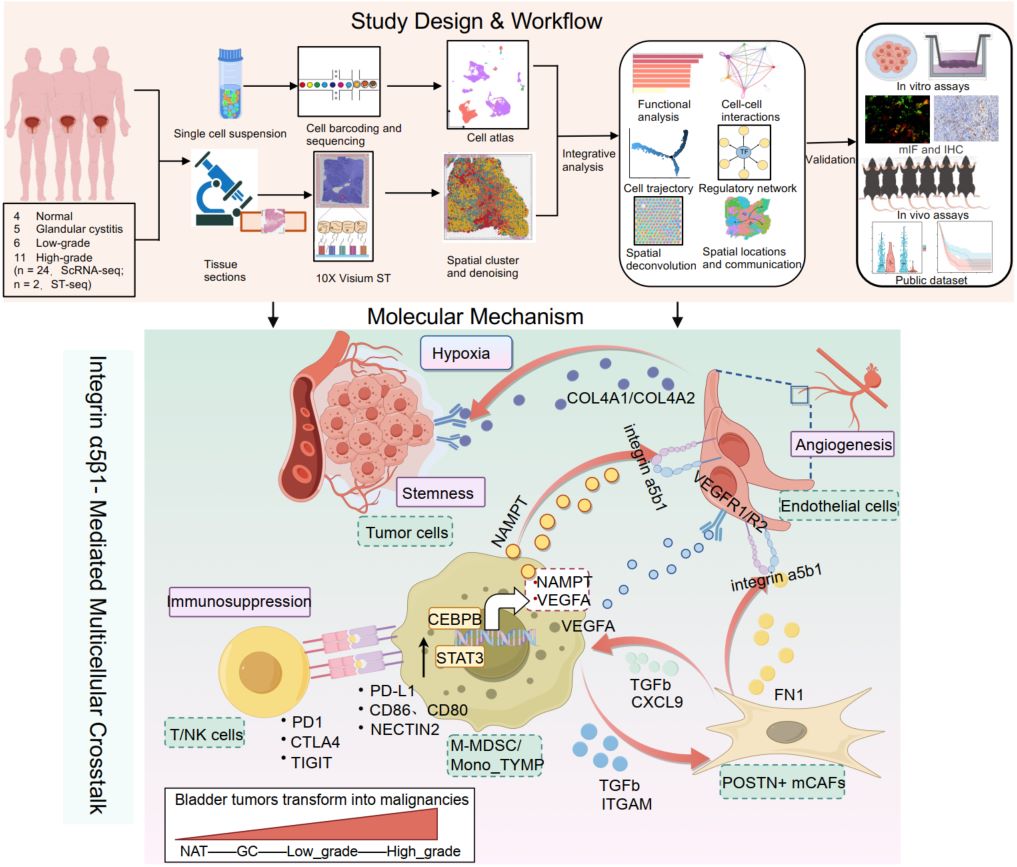
Integrating multiplexed immunofluorescence, animal models, and clinical samples, our single-cell and spatial atlas maps tumor microenvironment evolution during bladder cancer (BCa) progression. We reveal that stemness-associated tumor cells (SDC1+), POSTN+ myofibroblastic cancer-associated fibroblasts (mCAFs), and immunosuppressive monocytic myeloid-derived suppressor cells (M-MDSCs) are enriched in high-grade BCa. Mechanistically, mCAF-derived FN1 and M-MDSC-derived NAMPT engage endothelial integrin α5β1 to drive angiogenesis. Subsequently, endothelial cells sustain tumor stemness via the COL4A1/2–SDC1 axis. Therapeutically, targeting integrin α5β1 synergizes with immune checkpoint blockade, highlighting a promising stroma-directed strategy.
Pioneering the canine model in microbiota–gut–brain research for social and affective disorders
- 16 June 2026
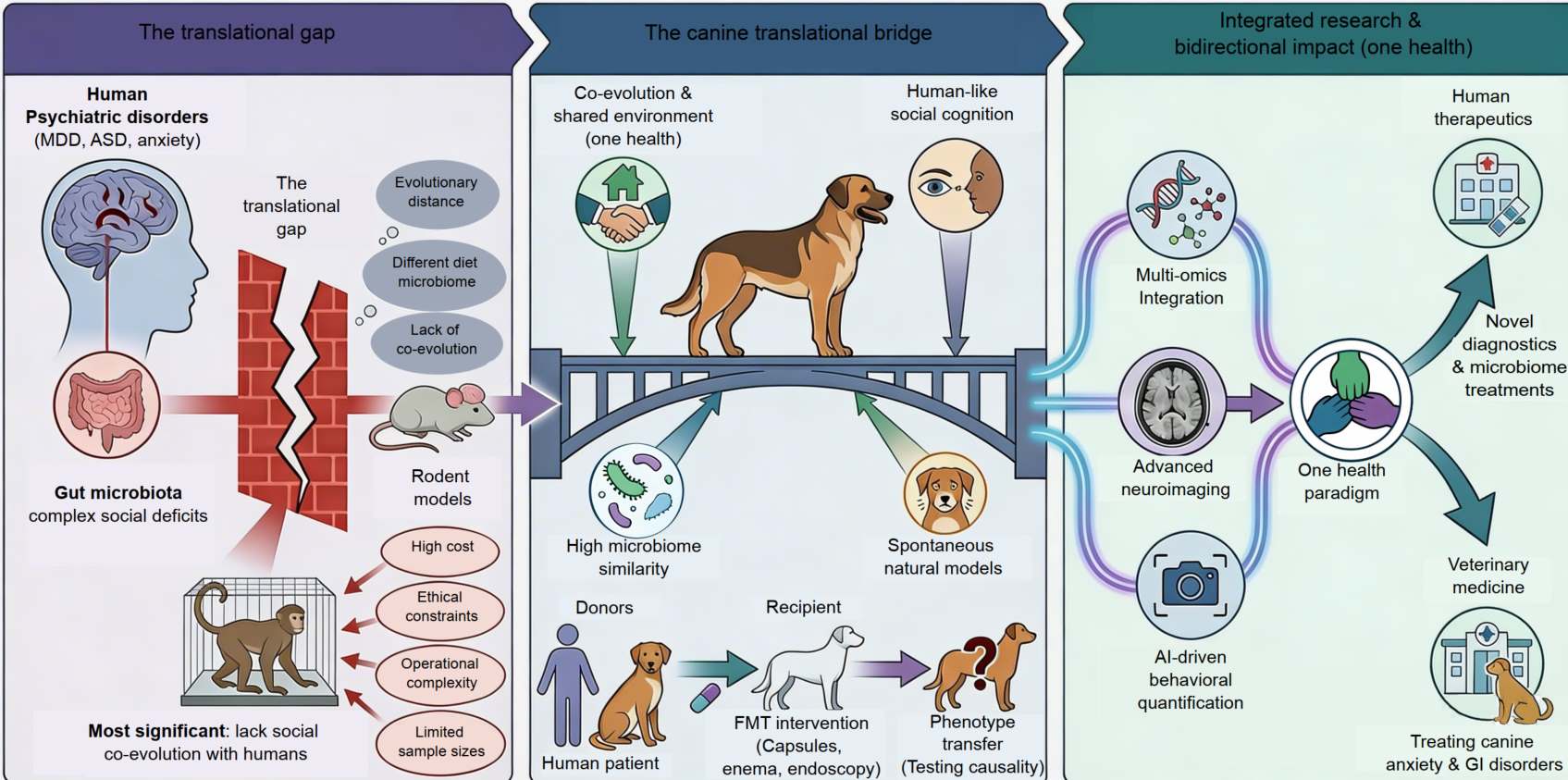
This commentary highlights the translational potential of canine models in investigating the microbiota–gut–brain (MGB) axis, focusing on their relevance to human social cognition and affective disorders, including major depressive disorder and autism spectrum disorder. While traditional rodent models provide foundational knowledge, the unique socio-behavioral complexity of dogs offers a potential bridge to study the shared neuroendocrine and microbial substrates underlying human psychopathology. This commentary outlines the promising avenues for canine-based research while critically addressing the practical limitations inherent in generalizing these findings to human populations, including genetic heterogeneity, standardization challenges, and ethical considerations. We propose a phased research pathway incorporating fecal microbiota transplantation and neurobehavioral phenotyping to define a clear trajectory for future translational research. Leveraging this canine platform creates a One Health paradigm, delivering mutual diagnostic and therapeutic advancements for both human and veterinary medicine, with applications for conditions such as anxiety and gastrointestinal disorders.
Majorbio Cloud: An integrated platform for single-omics and integrated multi-omics analysis of microbiome and metabolome
- 19 June 2026
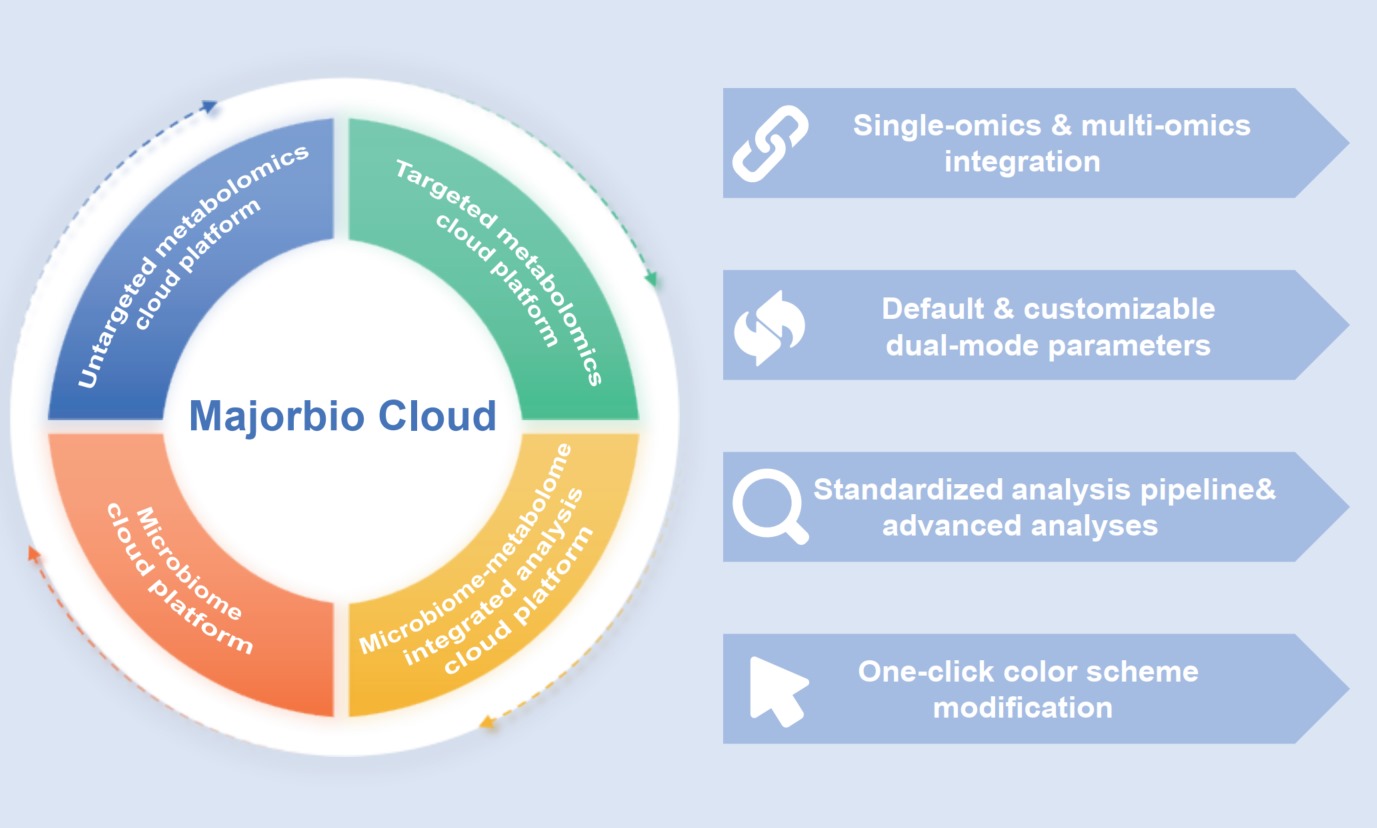
Advances in sequencing and mass spectrometry have propelled life sciences into the multi-omics era. Metabolomics and microbiomics are widely applied across various research fields, yet massive data analysis faces challenges like limited tool functionality and high technical barriers. The Majorbio Cloud Platform addresses these pain points by establishing four integrated workflows: Untargeted metabolomics, targeted metabolomics, microbiomics, and microbiome-metabolome integrated analysis, enabling one-stop analysis from single-omics to multi-omics correlation. In a real gut microbiome–metabolome case study, the platform pinpointed core differential microbes and characteristic metabolites, and further annotated functional pathways underlying host phenotypic divergence, demonstrating its reliability for mining biological mechanisms. The platform caters to both novice and experienced researchers with dual-parameter modes (default/customizable), multi-user collaboration, standardized pipelines, advanced analytical tools, and one-click color customization. By simplifying complex analytical procedures and reducing reliance on specialized programming skills, it offers a standardized, reproducible solution for multi-omics data analysis in related fields.
Gene expression signatures from single-cell transcriptomics predict Sjögren's syndrome
- 28 May 2026
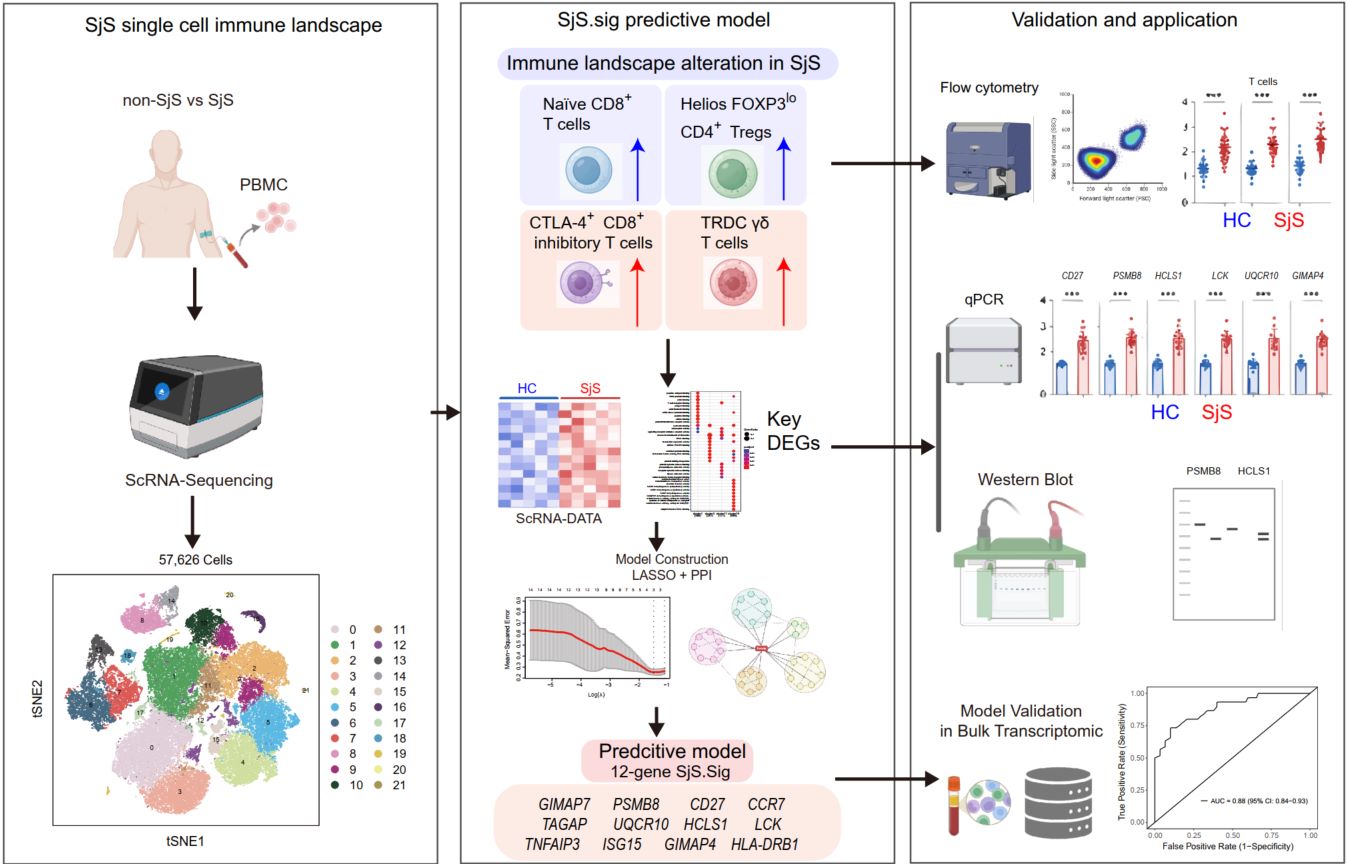
Sjögren's syndrome (SjS) is a systemic autoimmune disorder characterized by lymphocytic infiltration of exocrine glands, leading to dry eyes and mouth. While previous genome-wide association studies (GWAS) and transcriptomic analyses have identified genes associated with SjS, predictive models based on single-cell resolution are limited. In this study, single-cell RNA sequencing (scRNA-seq) data from peripheral blood mononuclear cells (PBMCs) of SjS patients were analyzed to map immune cell alterations linked to the disease. Compared with healthy controls, SjS patients displayed decreased proportions of naïve CD8+ T cells and Helios FOXP3lo CD4+ Tregs, alongside increased frequencies of CTLA−4+ CD8+ inhibitory T cells and TRDC γδ T cells. Using machine learning, a predictive model for SjS diagnosis was developed based on a 12-gene signature (SjS. Sig: GIMAP7, PSMB8, CD27, CCR7, TAGAP, UQCR10, HCLS1, LCK, TNFAIP3, ISG15, GIMAP4, and HLA-DRB1), which effectively differentiated patients from healthy individuals. Key genes such as CD27, PSMB8, HCLS1, LCK, UQCR10, and GIMAP4 were validated in clinical samples through flow cytometry and real-time quantitative PCR. These findings provide insights into the immune landscape of SjS at a single-cell resolution and propose a reliable molecular signature for diagnosis and immune monitoring.
Comprehensive spatial profiling reveals transitional microbiome dynamics and microbial heterogeneity in pediatric adenoid hypertrophy
- 10 May 2026
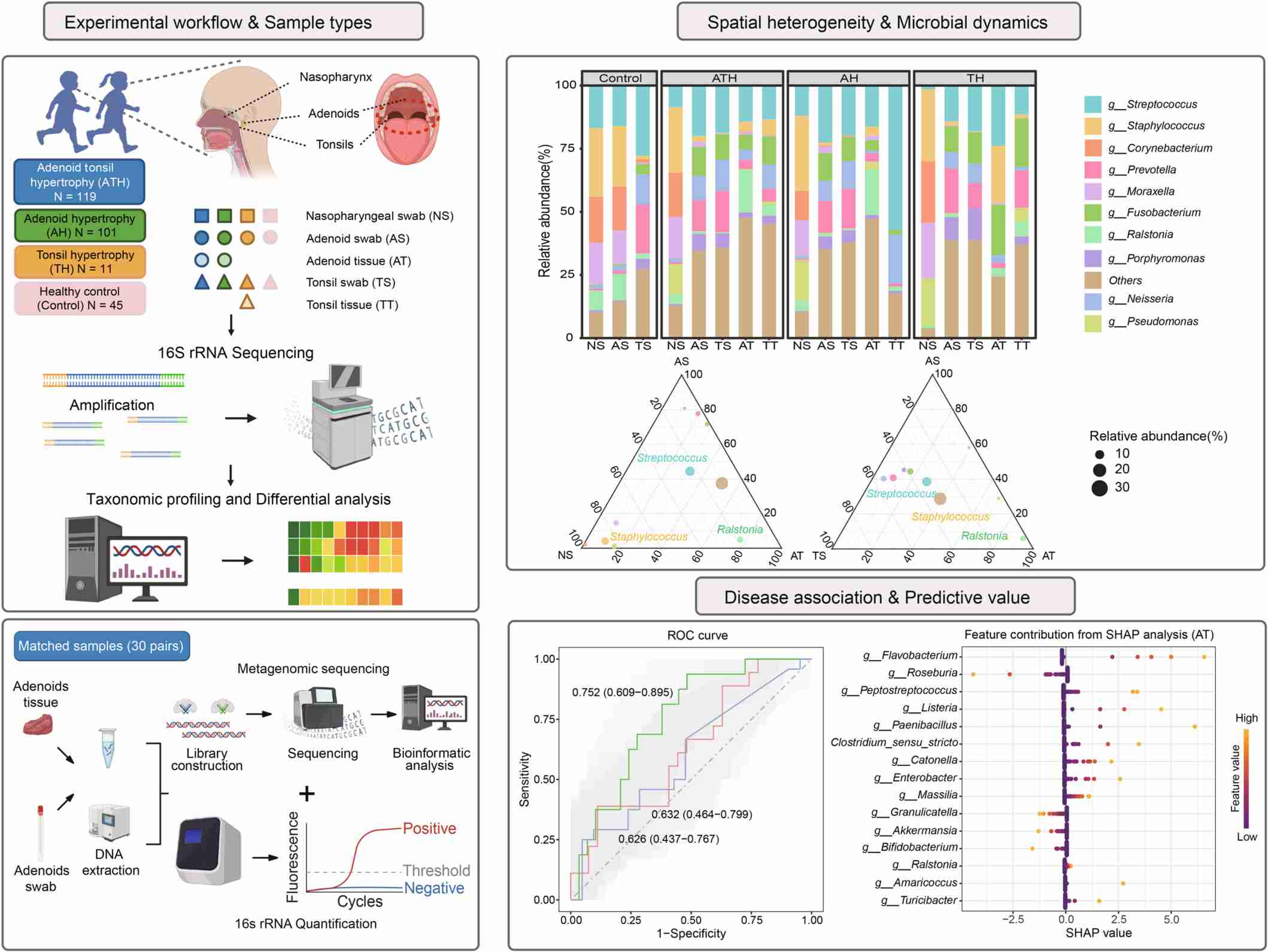
This study characterizes the upper respiratory microbiome in 276 children (101 Adenoid hypertrophy (AH) 119 Adenotonsillar hypertrophy (ATH), 11 Tonsil hypertrophy (TH), and 45 healthy controls by analyzing 1149 samples across five distinct niches: nasopharyngeal swabs (NS), adenoid swab (AS), and tonsil swabs (TS), plus adenoid tissues (AT) and tonsil tissues (TT). Utilizing a workflow of 16S rRNA sequencing, qPCR, and metagenomics, we identified significant spatial heterogeneity and niche-specific dominance, such as Staphylococcus in the nasopharynx and Streptococcus on mucosal surfaces. AT exhibited a transitional microbiome with intermediate composition and unique Ralstonia enrichment. Machine learning models demonstrate that AT microbial profiles (area under curve, AUC = 0.752) offer superior predictive power for AH severity compared to surface swabs. SHAP analysis identified key contributing genera, such as Roseburia and Flavobacterium, as primary drivers of these predictive outcomes. These findings highlight how tissue-infiltrating microbial dynamics drive chronic inflammation and pediatric hypertrophy.
Changes in the gut microbiome during fasting-induced molting and its effects on gut–liver function
- 20 May 2026
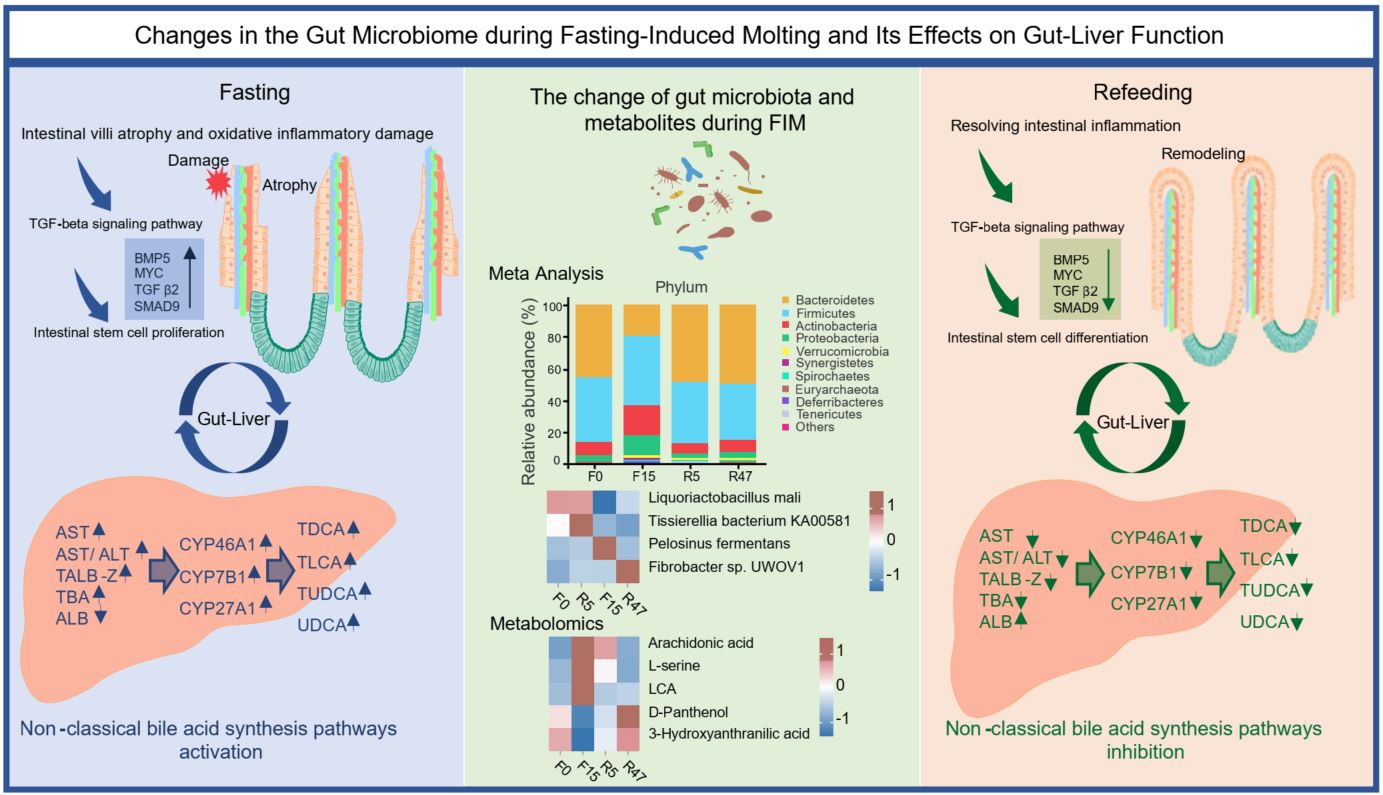
Fasting-induced molting (FIM) rejuvenates the laying cycle in hens; however, the fasting process may result in intestinal dysbiosis, which compromises host immunity and disrupts the intestine-liver function. Consequently, investigating the changes in gut microbiota during FIM and its impacts on gut-liver metabolic homeostasis is crucial for enhancing the efficiency of FIM in laying hens. Here, a total of 90 laying hens, aged 60 weeks, were selected for the FIM. Samples were collected at four time points: the day before fasting, the 15th day of fasting, the 5th day of refeeding, and the 47th day of refeeding. Metagenomic sequencing and non-targeted metabolomics were employed to investigate the roles of the gut microbiota and metabolites in the remodeling of gut–liver function, focusing on intestinal injury-repair mechanisms and changes in liver function. During the fasting period, the abundance of harmful microbiota and metabolites increase, leading to intestinal injury. This process activates the TGF-beta signaling pathway, promoting intestinal stem cell proliferation. Simultaneously, liver function dysfunction as evidenced by elevated bile acid levels in the liver and serum and activation of the non-classical bile acid synthesis pathway. During the refeeding period, the previously observed effects were reversed, leading to the remodeling of the intestinal microbiota and gut-liver function. We identified key microorganisms (Liquorilactobacillus mali and Tissierellia bacterium KA00581) and functional metabolites (d-Panthenol and 3-hydroxyanthranilic acid (3-HAA)) involved in intestinal-liver function during FIM. Notably, d-Panthenol promotes chicken small intestinal organoid branching and growth, enhances barrier function, and reduces inflammation. Our study underscores the role of gut microbiota in gut-liver injury during FIM in laying hens, suggesting potential probiotic- or metabolite-based interventions to mitigate gut-liver injury and facilitate recovery.
Genomic epidemiology and lineage-specific risk stratification of tet(X4)-mediated tigecycline resistance along the pork production chain: A One Health perspective
- 20 May 2026
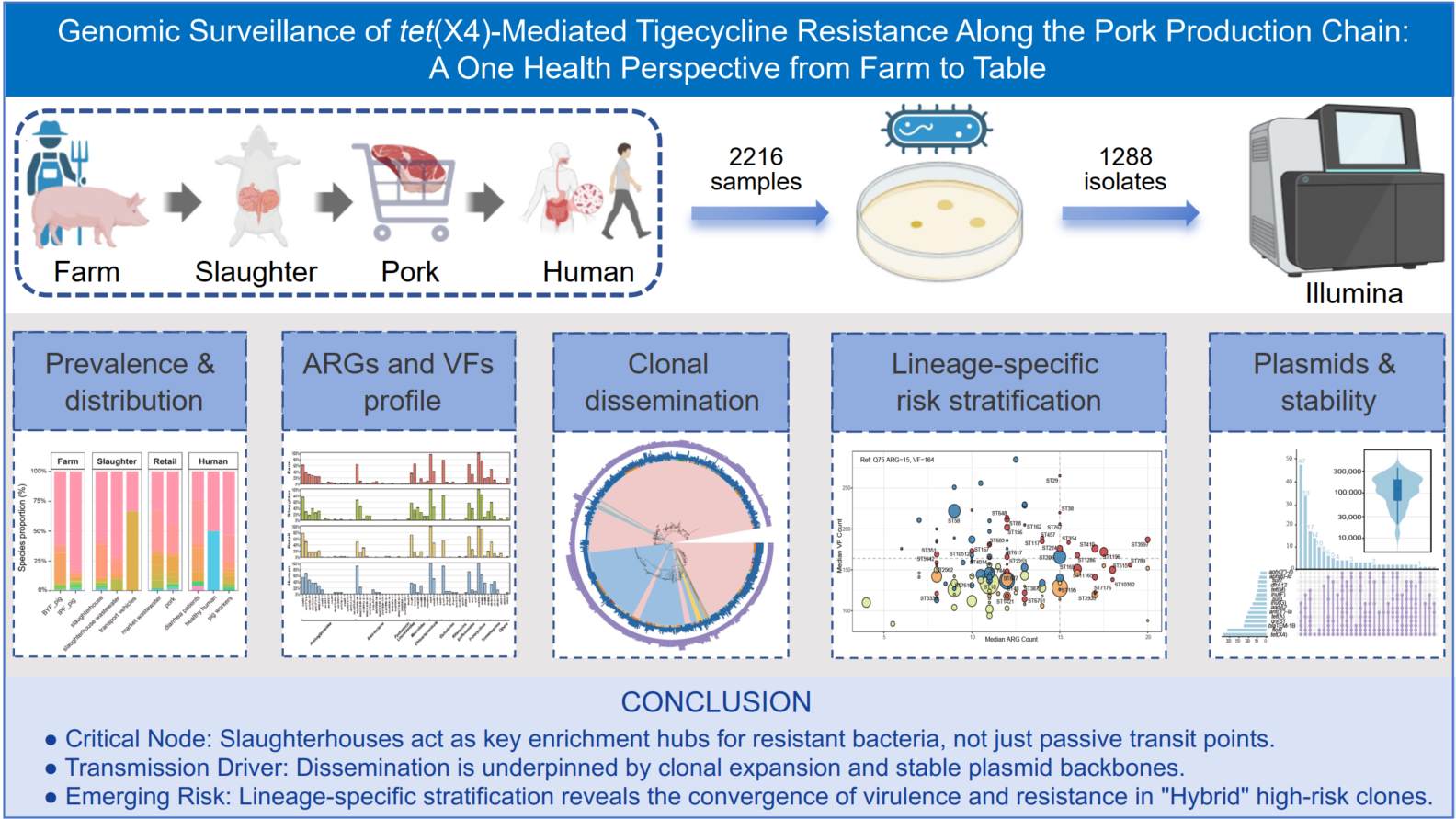
Tigecycline serves as a last-resort antibiotic, yet its efficacy is increasingly compromised by the rapid dissemination of the plasmid-borne resistance gene tet(X4). While metagenomics has broadened our understanding of resistomes, high-resolution genomic tracking of resistant clones across the entire farm-to-table continuum remains critical. In this study, we conducted a large-scale surveillance project in China, analyzing 2216 samples across the pork production chain and isolating 1288 tigecycline-resistant strains. Our genomic analysis revealed a distinct transmission dynamic: while intensive pig farms exhibited the highest recovery rate under selective enrichment, slaughterhouses acted as a critical enrichment node and served as a reservoir and potential source for the introduction of resistant strains into downstream food systems. Notably, occupational exposure emerged as an important exposure pattern, with pig farm workers showing markedly higher carriage rates than the general population. Whole-genome sequencing of 790 tet(X4)-positive Escherichia coli isolates revealed a population structure dominated by phylogroups A and B1. ST10 and ST195 were the most prevalent lineages and showed extensive putative clonal relatedness across the farm-slaughterhouse interface. We identified 2574 putative clonal relatedness events (SNPs ≤ 10), heavily concentrated in the farm-slaughterhouse interface. To assess public health threats, we developed a lineage-specific risk stratification model by integrating global datasets. This revealed two parallel risk trajectories: the enrichment of multidrug-resistant commensals in processing nodes and the convergence of hyper-virulence and resistance in specific Hybrid lineages. Furthermore, analysis of 234 tet(X4)-bearing plasmids elucidated a conserved ISVsa3-rdmC-tet(X4) genetic module carried primarily by IncF/IncHI/IncX1 backbones. We further uncovered that the stability of these plasmids is underpinned by lineage-specific Toxin-Antitoxin systems, specifically the double-lock mechanism (RelBE and Phd/Doc) in dominant IncX1 plasmids. These findings highlight farms and slaughterhouses as pivotal control points and demonstrate the power of integrated genomic surveillance in guiding One Health strategies to mitigate the spread of tet(X4).
Integrated high-throughput phenomics and transcriptomics uncover the transcriptional mechanisms underlying rice responses to elevated CO2 concentration and cadmium stress
- 20 May 2026
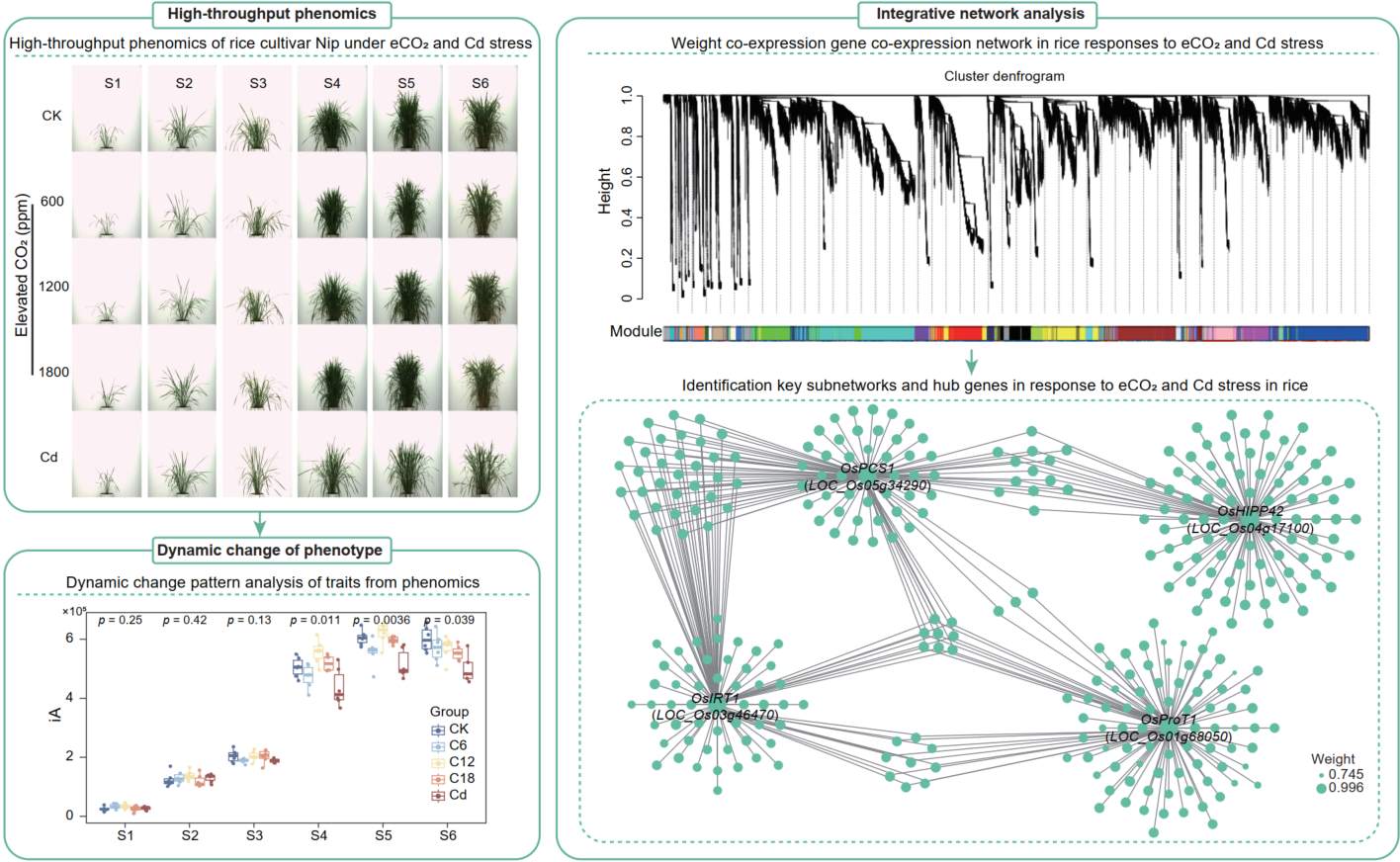
Developing crop varieties with reduced cadmium (Cd) accumulation under elevated carbon dioxide (eCO2) is a major global challenge. Identifying hub genes and elucidating the transcriptional mechanisms governing Cd stress and eCO2 responses requires integrated, multi-tissue omics approaches. Here, we conducted high-throughput phenotyping of the rice cultivar Nipponbare across six key developmental stages under Cd stress and three eCO2 concentrations. Image-based traits revealed shared and distinct phenotypic responses to these stresses. Multi-tissue transcriptome analyses showed that the differentially expressed genes (DEGs) and hormone-related genes exhibited a similar expression pattern under Cd and eCO2. We further constructed an integrative co-expression network and identified five stress-responsive subnetworks. Among these, Multiple Stress Responsive Gene 3 (OsMSR3) emerged as a hub gene, with its haplotype 2 (Hap2) frequency decreasing from low- to high-latitude regions and from southern to northeastern China, mirroring the geographic distribution of Cd-contaminated soils. Collectively, our study reveals a transcriptional regulatory network underlying rice responses to Cd and eCO2 stresses, providing mechanistic insights and valuable genetic resources for future molecular breeding of low-Cd accumulating rice varieties adapted to constant rising CO2 levels.
Engineered Bacillus subtilis regulates intestinal barrier in ETEC K88-infected piglets and modulates miR-1250-5p/NFκB pathway in IPEC-J2
- 11 May 2026
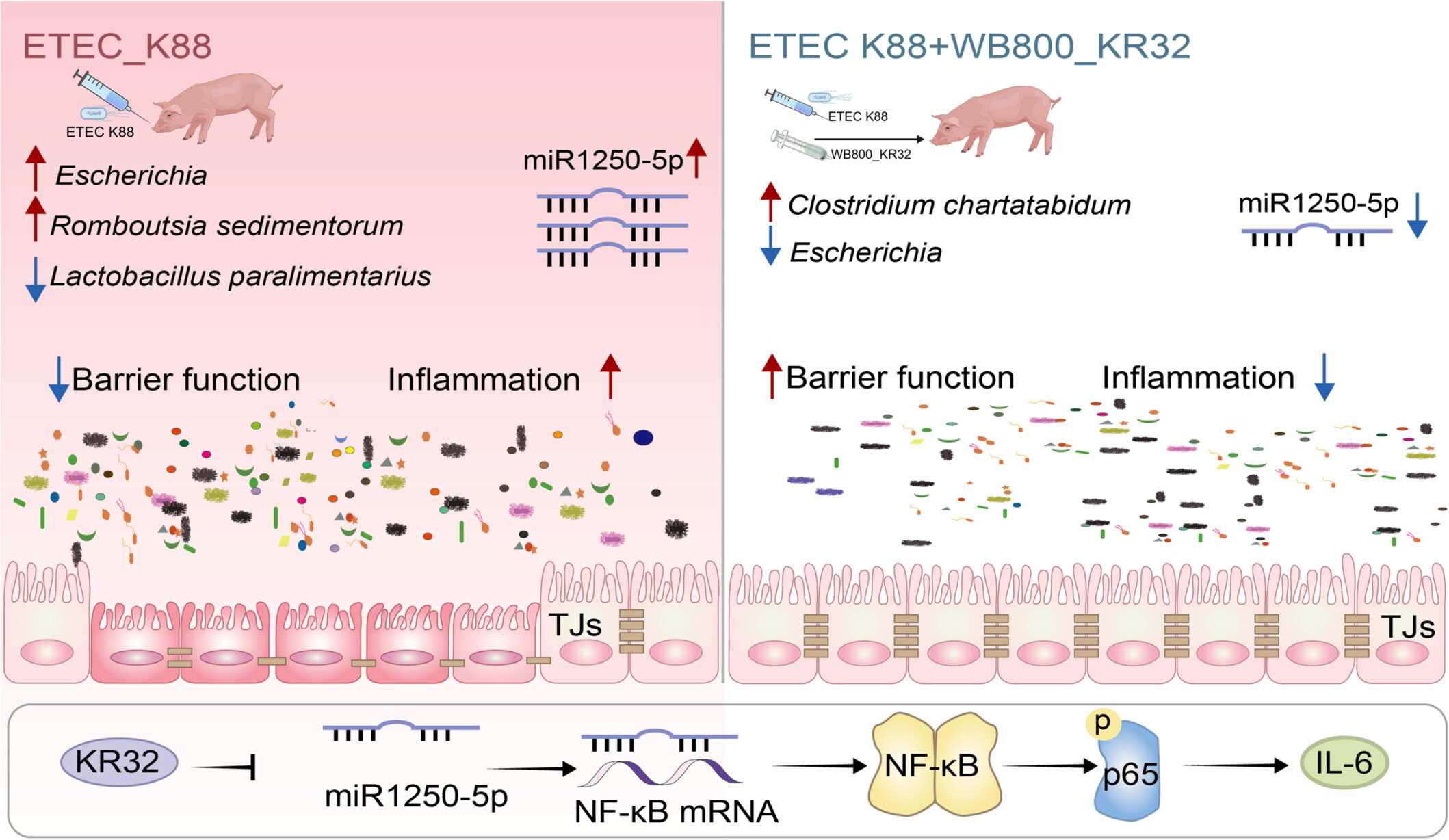
Upon enterotoxigenic Escherichia coli (ETEC) K88 infection, intestinal barrier function is disrupted with compromised tight junctions, accompanied by an exacerbated inflammatory response, proliferation of pathogenic Escherichia, and depletion of beneficial Lactobacillus paralimentarius. In parallel, miR-1250-5p expression is elevated, and both NF-κB mRNA and phosphorylated p65 protein levels are increased. With WB800-KR32 pretreatment followed by ETEC K88 challenge, KR32 antimicrobial peptides are secreted to directly inhibit Escherichia, intestinal barrier integrity is restored with reduced inflammation, the microbiota is reshaped with increased Lactobacillus paralimentarius, decreased Escherichia, and altered abundances of Romboutsia sedimentorum and Clostridium chartatabidum, miR-1250-5p expression is downregulated, and NF-κB signaling is suppressed, as evidenced by reduced NF-κB mRNA levels and diminished p65 phosphorylation. Central Mechanism: The engineered bacterium employs a two-pronged strategy: modulating the miR-1250-5p/NF-κB pathway and delivering KR32 antimicrobial peptide to improve gut microbiota composition, strengthen intestinal barrier function, and mitigate ETEC K88-induced inflammatory damage.
The mediating roles of gut microbiota in the associations of air pollution and meteorological factors with metabolic syndrome
- 07 May 2026
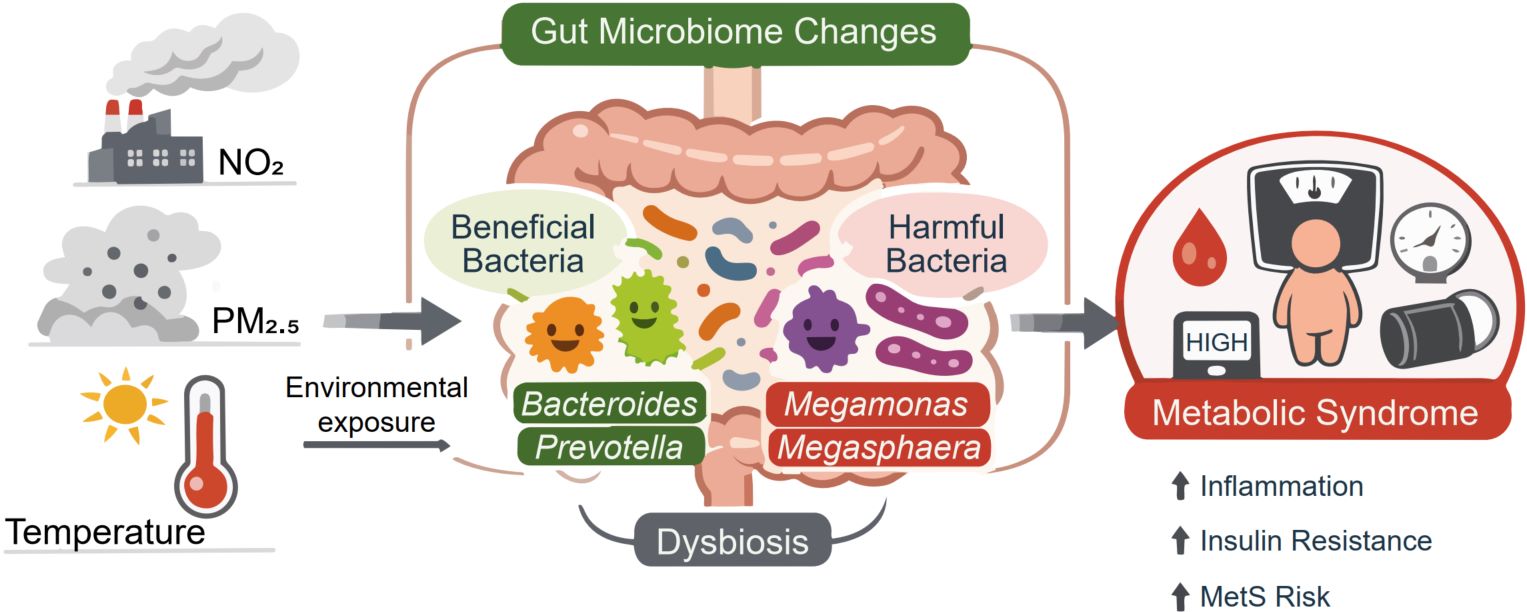
We characterized distinct gut microbial community clusters associated with metabolic syndrome (MetS) and identified key genera underlying these patterns. Health-associated clusters were dominated by beneficial genera such as Bacteroides and Prevotella with known metabolic protective functions. Environmental exposures (air pollution and temperature) were associated with gut microbiota composition, and may increase the risk of MetS. Our findings further suggest that specific bacterial genera, particularly Megamonas and Megasphaera, may statistically mediate the associations of long-term exposure to nitrogen dioxide (NO2), fine particulate matter (PM2.5), and ambient temperature with MetS risk. Integrating environmental exposure assessment with gut microbiota profiling offers a promising strategy for metabolic syndrome prevention and precision intervention.
L-EasyARG: A long-read metagenomics tool for rapid antibiotic-resistance-gene profiling
- 21 April 2026
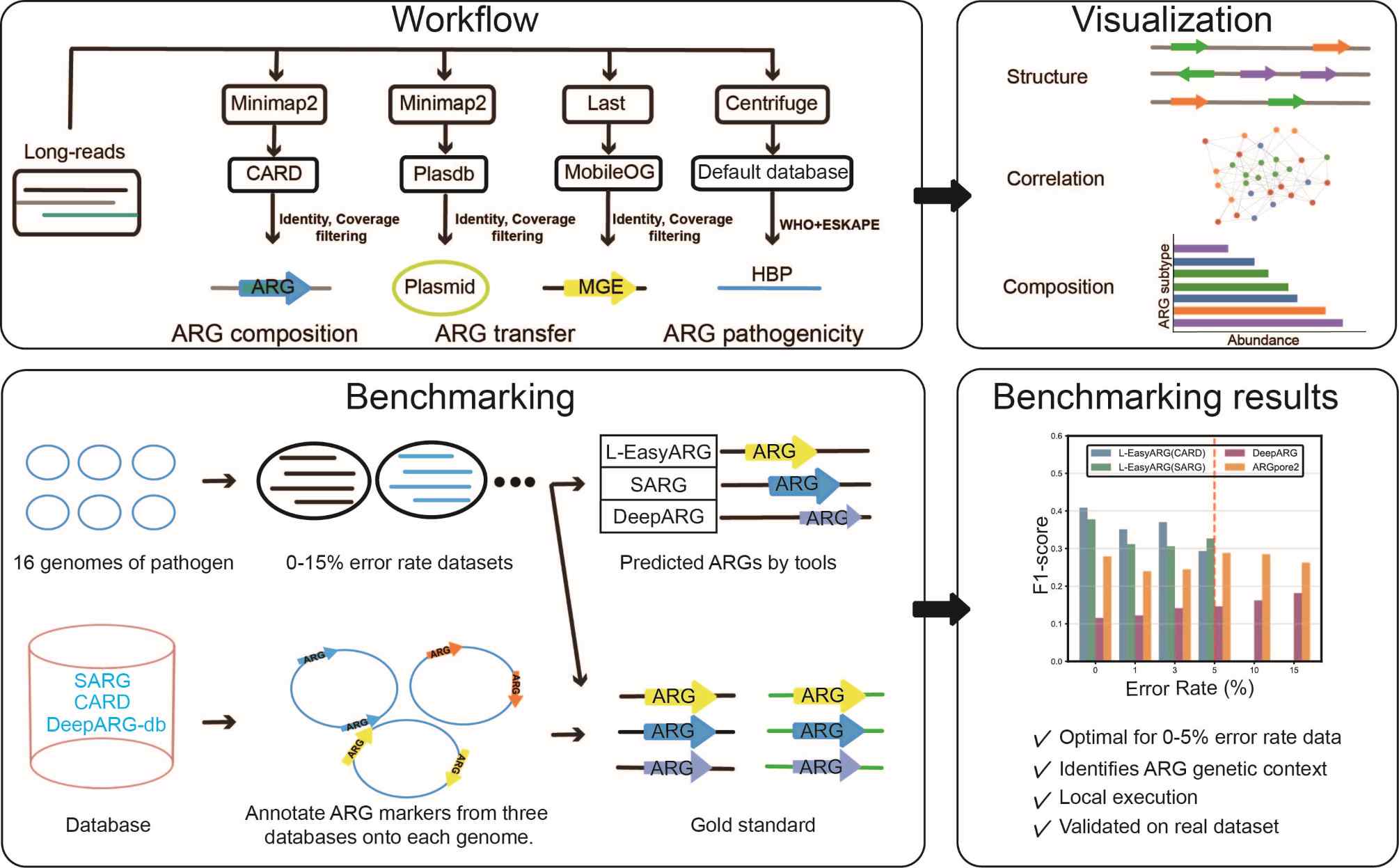
L-EasyARG is a pipeline for long-read metagenomic profiling of antibiotic resistance genes (ARGs) and their genetic context. It integrates ARG identification, plasmid and mobile genetic element (MGE) detection, taxonomic classification, and further identifies ARGs carried by human bacterial pathogens (HBPs) defined in the WHO and ESKAPE pathogen lists. Benchmarking on simulated datasets with error rates ranging from 0% to 15% demonstrates optimal F1-scores at ≤5% errors, outperforming existing tools such as ARGpore2 and DeepARG. Application to hospital wastewater samples validates its ability to capture disinfection-induced shifts in ARG composition, ARG-MGE co-occurrence, and ARG-host associations. L-EasyARG generates comprehensive outputs including abundance profiles, co-occurrence networks, and interactive visualizations, enabling efficient resistome surveillance and risk assessment. The pipeline is user-friendly and locally executable.
Beyond “you are what you eat”: Unlocking gut microbiota-mediated biotransformation of dietary phytochemicals
- 05 April 2026
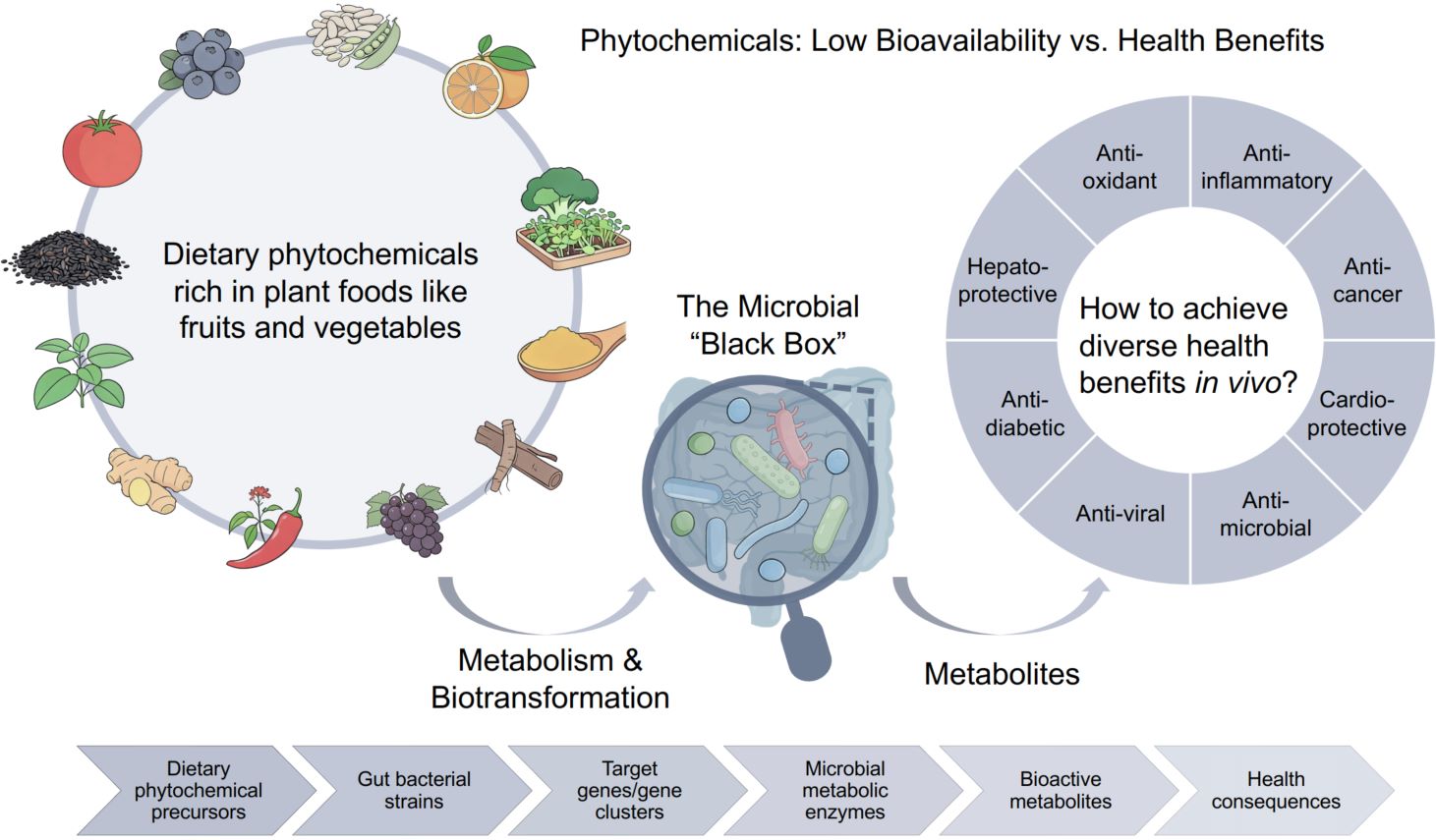
The “efficacy paradox” of phytochemicals, low bioavailability yet significant health benefits, is associated with gut microbiota, which biotransforms dietary precursors into bioactive metabolites, enabling systemic effects. Thus, health outcomes of diet depend not just on intake “the rainbow”, but on gut microbial metabolism, redefining “you are what you eat” to “you are what your microbes make of what you eat”.
Pangenome variation analysis uncovers the impacts of SNP-independent structural variations in modern European cattle breeding
- 05 April 2026
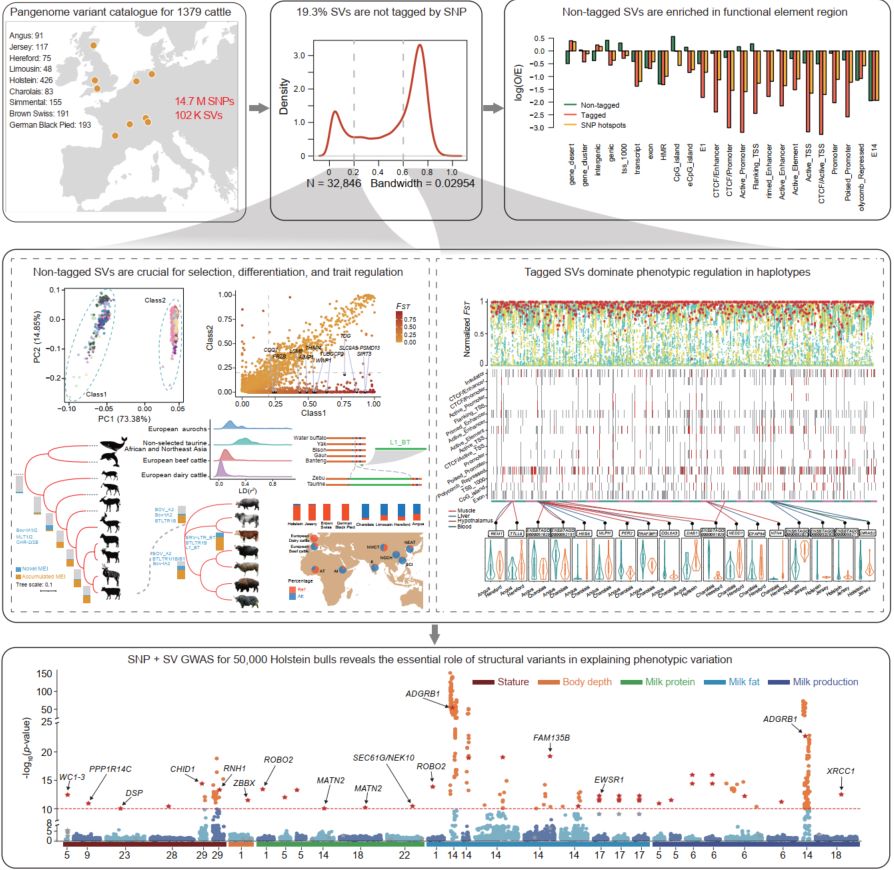
This study built a pan-genome single-nucleotide polymorphism (SNP) + structural variations (SVs) map for 1379 European cattle. It revealed that SVs not tagged by SNPs are crucial for selection, differentiation, and trait regulation, whereas SNP-tagged SVs dominate phenotypic regulation within haplotypes, collectively proving SVs' essential role in explaining phenotypic variation.
CGAS (Chloroplast Genome Analysis Suite): An automated python pipeline for comprehensive comparative chloroplast genomics
- 31 March 2026
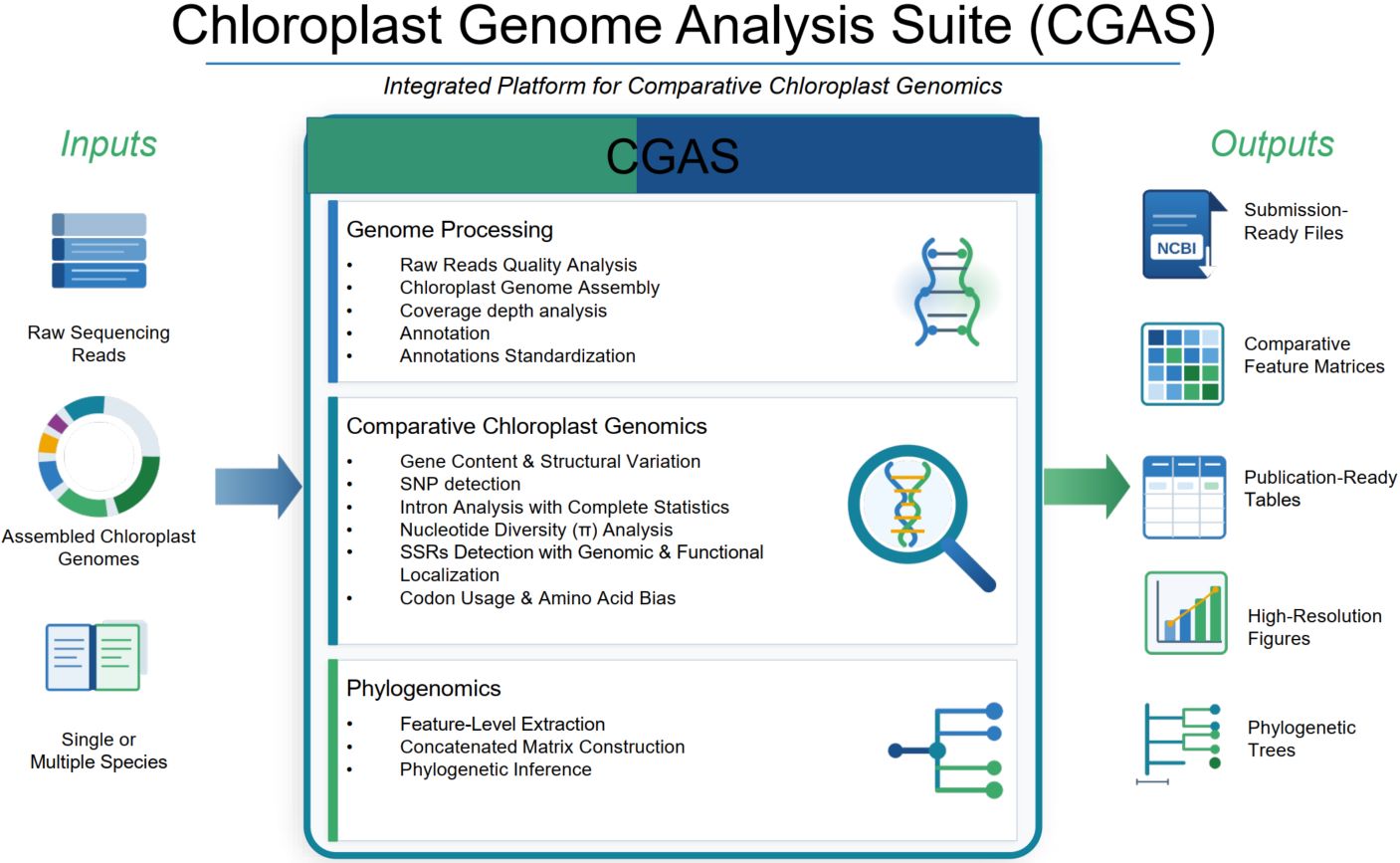
Chloroplast Genome Analysis Suite (CGAS) is a comprehensive bioinformatics pipeline that streamline chloroplast genome analysis from raw sequencing reads to publication-ready outputs. The suite integrates 14 specialized modules organized across three sequential phases. Phase 1 (Modules 1–4) handles genome assembly, quality control, annotation, gene normalization, and National Center for Biotechnology Information-format conversion using tools such as fastp, GetOrganelle, PGA, and others. Phase 2 (Modules 5–13) enables batch comparative genomics, encompassing gene content comparison, genome structure characterization, codon usage analysis, amino acid composition, single-nucleotide polymorphism detection, intron profiling, simple sequence repeat identification, and nucleotide diversity assessment—with R-based visualizations integrated where graphical representation of results is required. Phase 3 (Module 14) performs robust phylogenetic inference via alignment and tree-building programs, including MAFFT, MACSE, IQ-TREE, and others. Together, CGAS transforms raw chloroplast genome data into comparative insights, providing researchers with an automated, reproducible, and scalable solution for comparative chloroplast genomics, available at https://github.com/abdullah30/Chloroplast-Genome-Analysis-Suite-CGAS.
Ginseng polysaccharides prevent mastitis through Lactobacillus murinus-derived deoxycholic acid and TGR5 signaling
- 03 March 2026
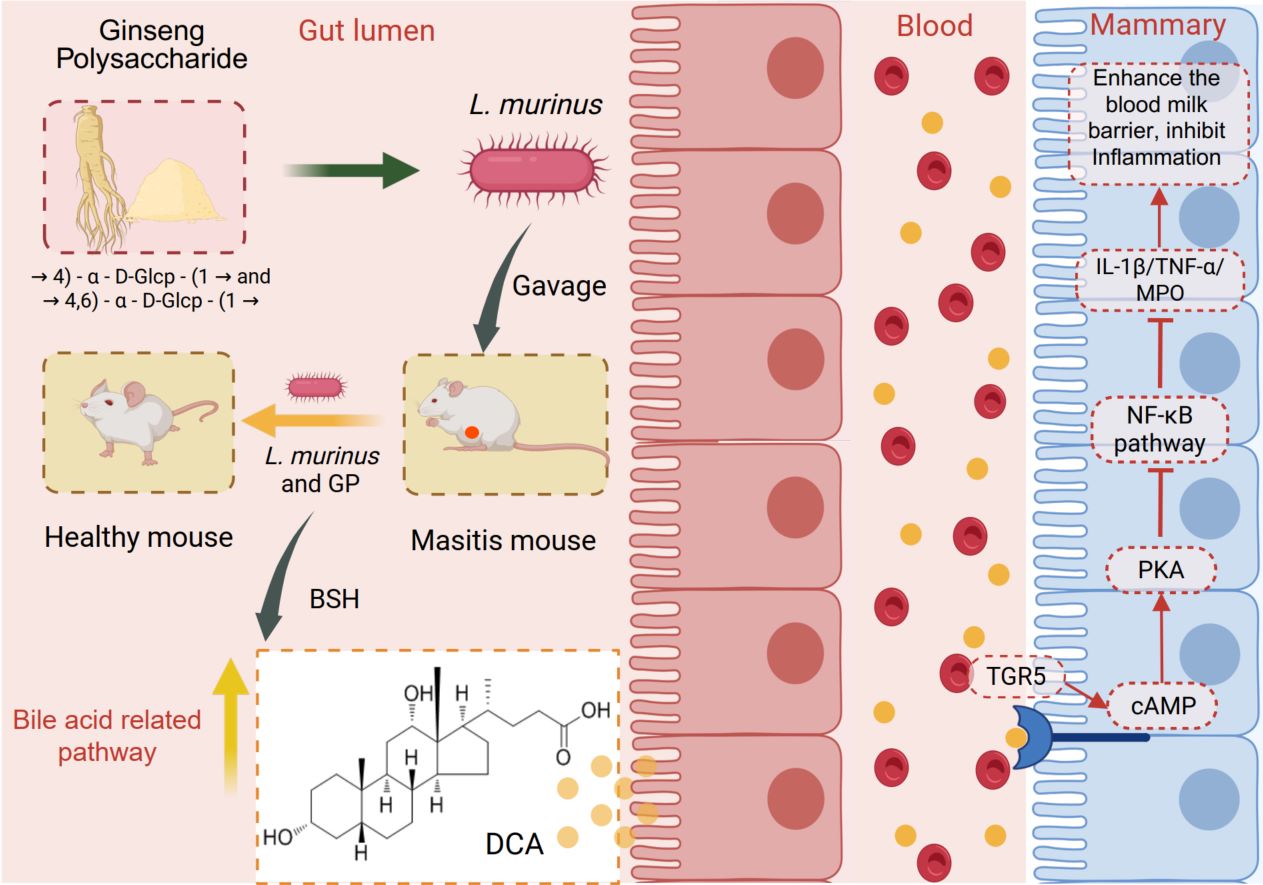
Prebiotic Ginseng polysaccharides (GP) alleviate mastitis through selective enrichment of gut L. murinus, which elevates its anti-inflammatory metabolite deoxycholic acid (DCA). Circulating DCA engages mammary epithelial TGR5 receptors, triggering the cAMP–PKA pathway to suppress NF-κB/NLRP3-mediated inflammation. The synergistic action of GP and L. murinus underscores the translational promise of this synbiotic strategy. Targeting the gut–mammary axis—specifically the identified microbial (L. murinus) and metabolic circuits—offers a compelling, non-antibiotic approach for mastitis prevention and therapy.
Functionally complementary bacterial inoculant coordinates arbuscular mycorrhizal fungi to improve Angelica sinensis root yield and quality
- 14 March 2026
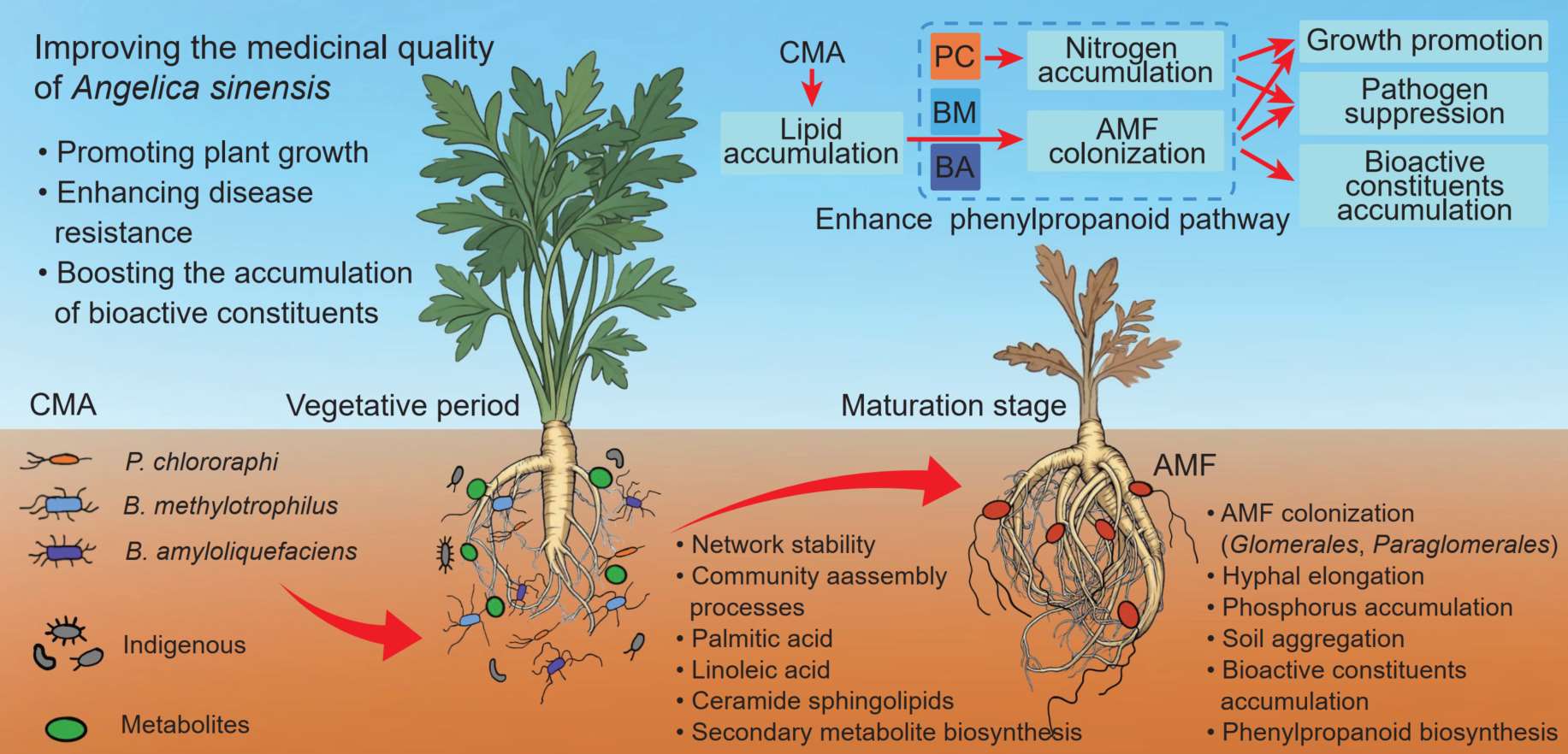
Comprehensive understanding of how diverse PGPR strains enhance the rhizosphere microenvironment remains a considerable challenge. Here, we provide experimental evidence that a functionally synergistic composite microbial formulation can markedly enhance growth performance and improve the quality attributes in Angelica sinensis. Our findings present a practical strategy for simultaneously controlling soil-borne diseases and boosting the medicinal value of root- and rhizome-based medicinal plants.
Arbuscular mycorrhizal fungal community abundance, functions, and symbiotic interactions revealed by root metatranscriptomes
- 21 February 2026
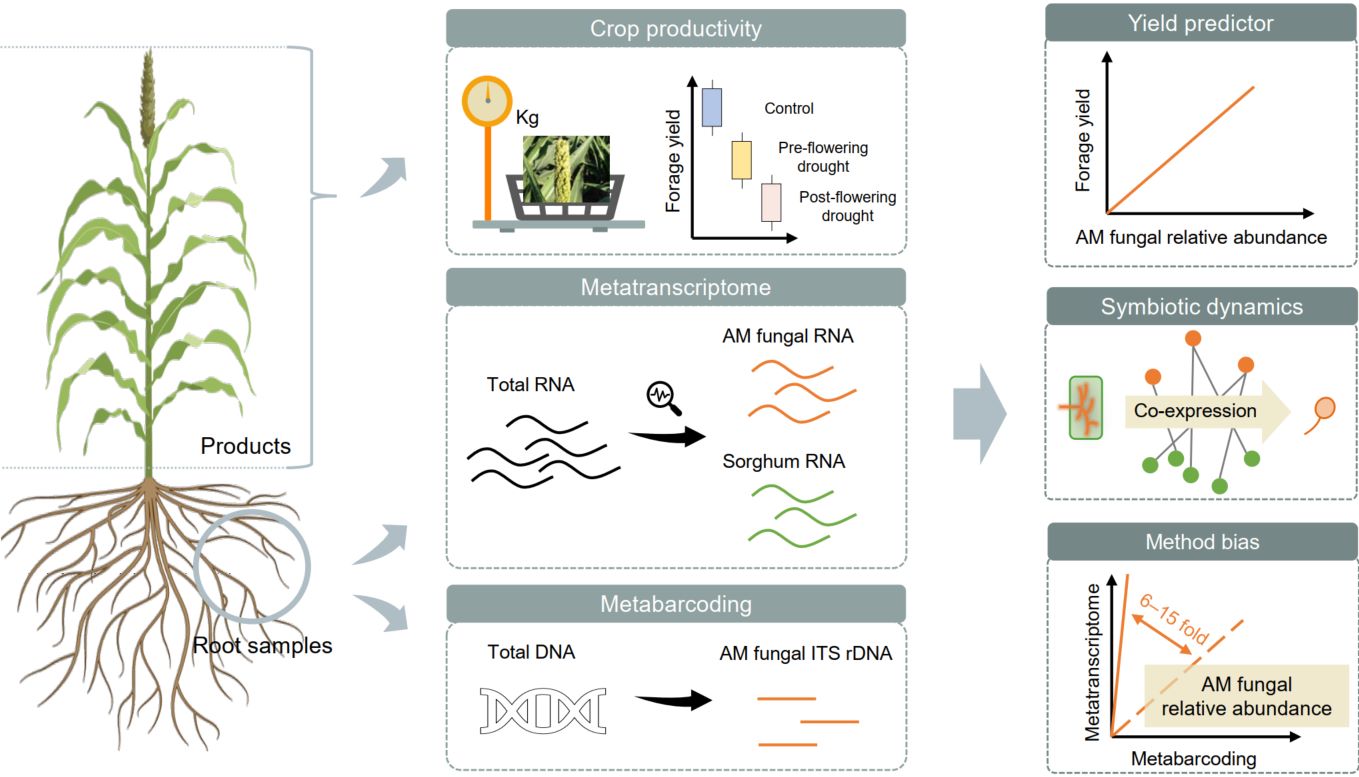
Paradigm shift: PCR-free methods reveal 6–15-fold higher arbuscular mycorrhizal (AM) fungal abundance than metabarcoding, exposing systematic underestimation across decades of research. Predictive power: AM fungal abundance serves as a community-level trait that predicts crop yield under drought conditions. Symbiotic dynamics: Symbiotic life history from arbuscule formation to sporulation is governed by coupled and decoupled co-expression of plant and AM fungal genes.
Stratification of chronic rhinosinusitis with nasal polyps by distinct sinonasal microbial communities and their clinical and pathological correlations
- 06 January 2026
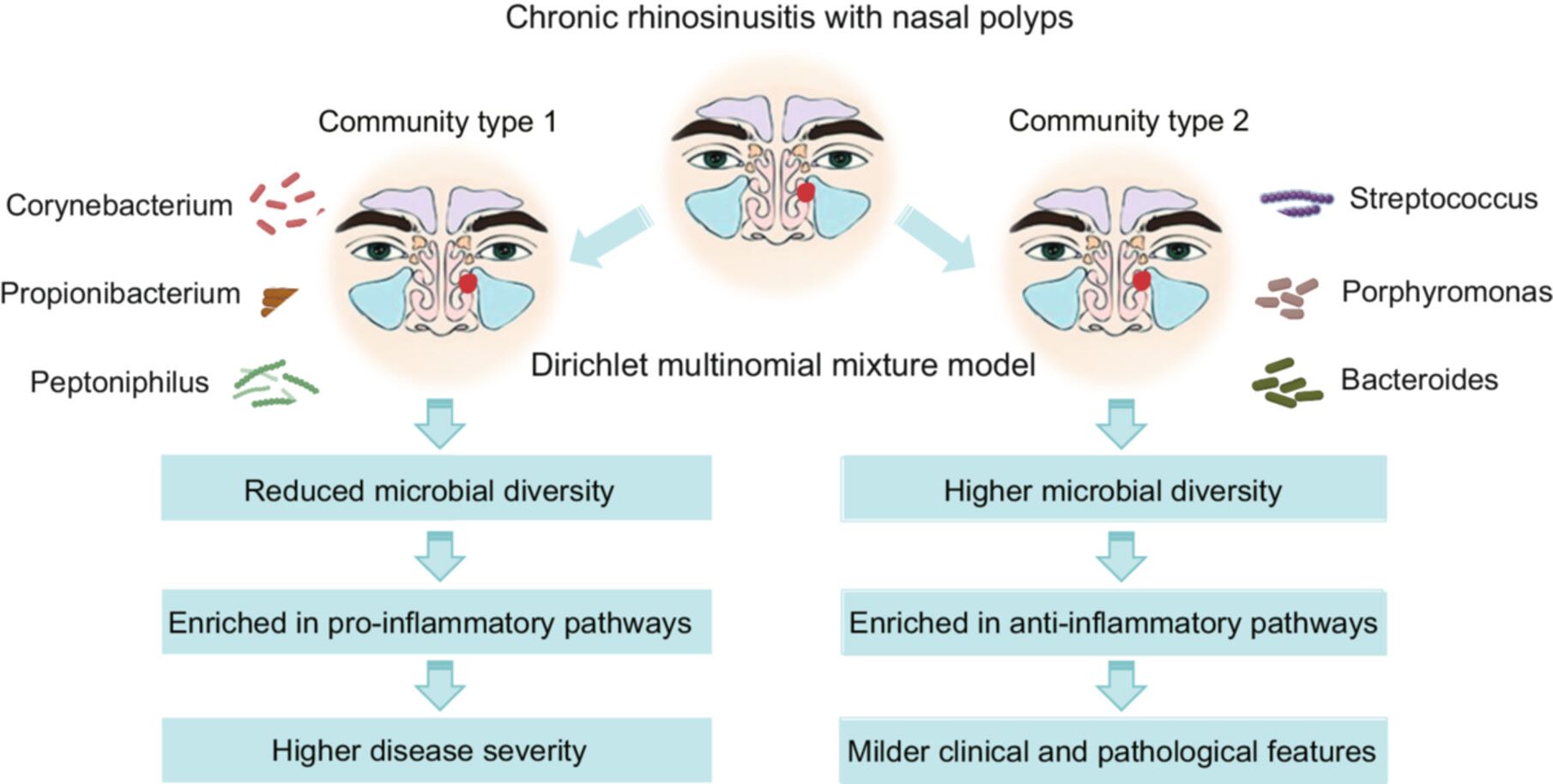
Chronic rhinosinusitis with nasal polyps (CRSwNP) is a heterogeneous inflammatory condition. Dysbiosis is commonly observed. This study identified distinct sinonasal microbial communities in CRSwNP patients and explored their clinical and pathological associations. Two distinct microbial communities were identified: community type 1 (CT1), dominated by Corynebacterium, Propionibacterium, and Peptoniphilus; and community type 2 (CT2), dominated by Streptococcus, Porphyromonas, and Bacteroides. CT1 exhibited significantly reduced microbial diversity and was enriched in pro-inflammatory pathways, whereas CT2 showed higher microbial diversity, with pathways supporting anti-inflammatory. Clinically, CT1 patients demonstrated higher disease severity and increased eosinophil infiltration, compared with CT2. CRSwNP patients can be stratified into two distinct microbial communities, each with specific clinical and pathological profiles. Microbial-based stratification may inform tailored therapeutic strategies to address the diverse clinical and pathological manifestations of CRSwNP.
Microbial influences on immune modulation and colorectal cancer progression through combined transcriptomic and microbiomic analysis
- 04 January 2026
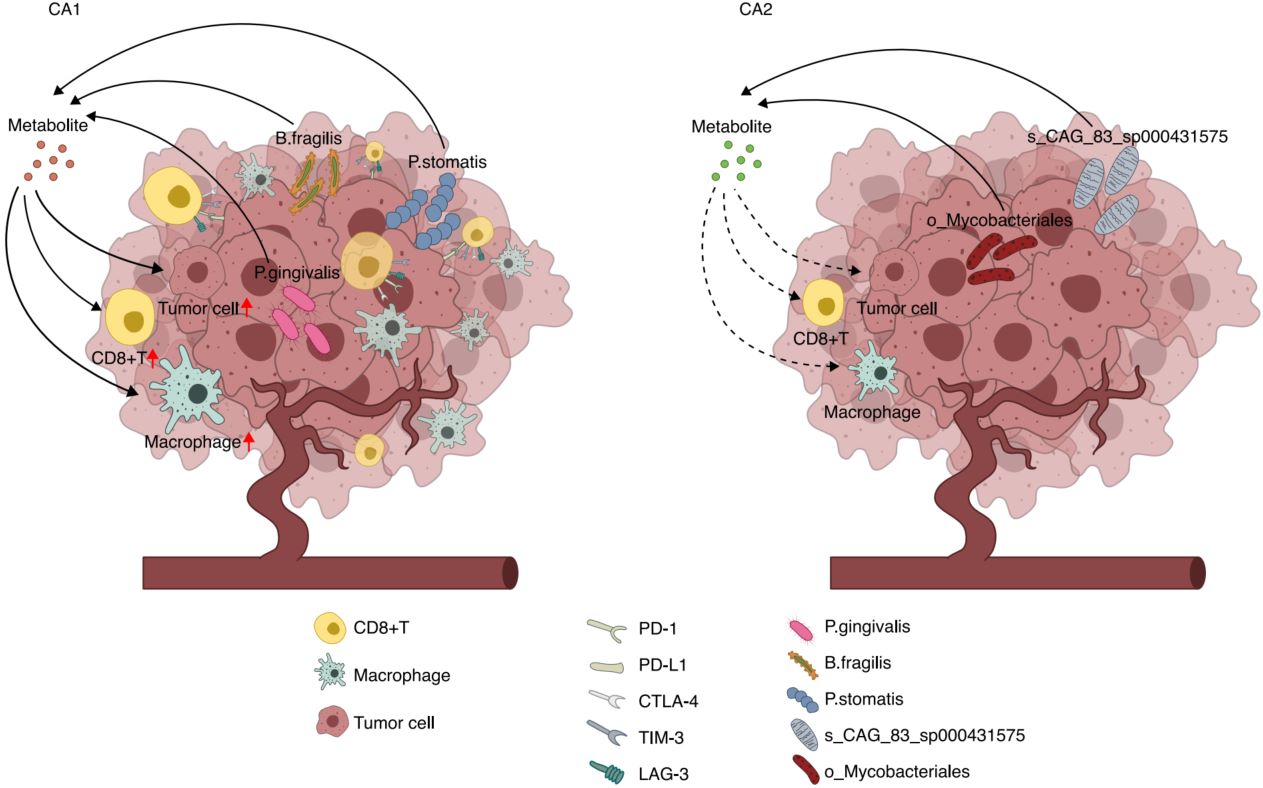
Schematic representation of the distinct tumor microenvironment characteristics in CA1 and CA2 subtypes. CA1 illustrates an immune-active tumor microenvironment characterized by elevated infiltration of immune cells, particularly CD8+T cells and macrophages, and enhanced IFN-γ signaling. This subtype shows enrichment of specific bacterial species (including P. gingivalis and B. fragilis) that contribute to immunomodulation through metabolic pathways such as γ-aminobutyric acid (GABA) and butanoate metabolism. Despite high immune activity, CA1 exhibits a poor prognosis, potentially linked to immune exhaustion, manifested by high expression of PD-1, CTLA-4, LAG-3, and TIM-3. CA2 depicts an immune-suppressed microenvironment featuring reduced immune cell infiltration, lower inflammatory signatures, and a distinct microbial composition dominated by health-associated bacteria. This tumor phenotype shows better overall survival even with reduced immune activity. Arrows indicate key molecular interactions, with solid lines representing direct effects and dashed lines indicating indirect modulation.
The gut microbiome promotes the growth performance of black soldier fly larvae by detoxifying uric acid
- 24 December 2025
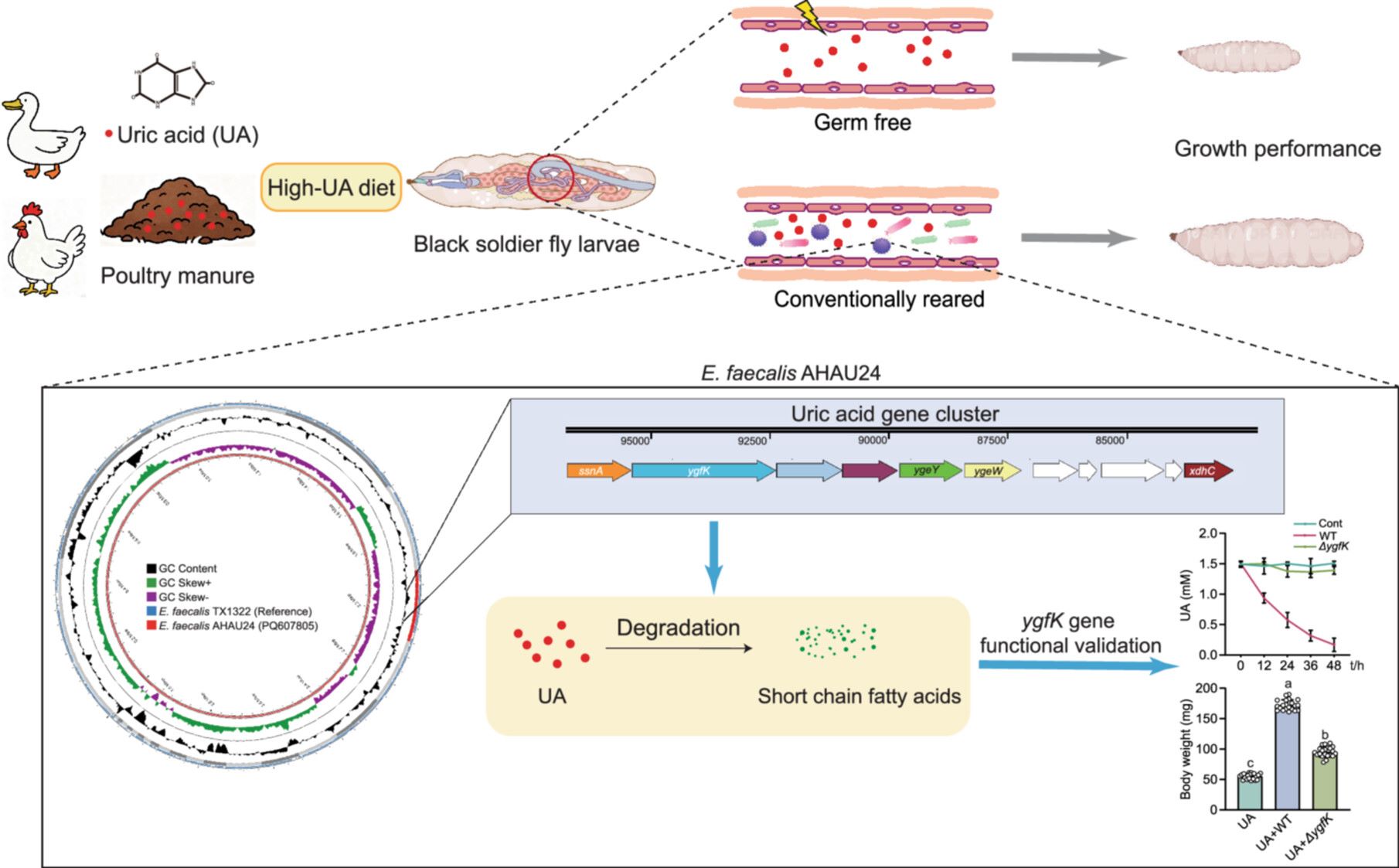
This study demonstrates the detrimental effects of exogenous uric acid (UA) on the growth of black soldier fly (BSF) larvae, highlighting the role of gut microbiota in UA degradation. We isolated UA-degrading bacterial strains associated with BSF, including Enterococcus faecalis AHAU24. Genomic analysis identified a UA degradation gene cluster in this strain, providing insights into its potential for UA catabolism. Furthermore, mono-association of germ-free larvae with E. faecalis AHAU24 confirmed that this strain alleviates UA-induced growth retardation and enhances bioconversion efficiency by degrading dietary UA.
MicrobTiSDA: A flexible R package for inferring interspecies interactions and abundance dynamics in microbiome time-series data
- 27 November 2025
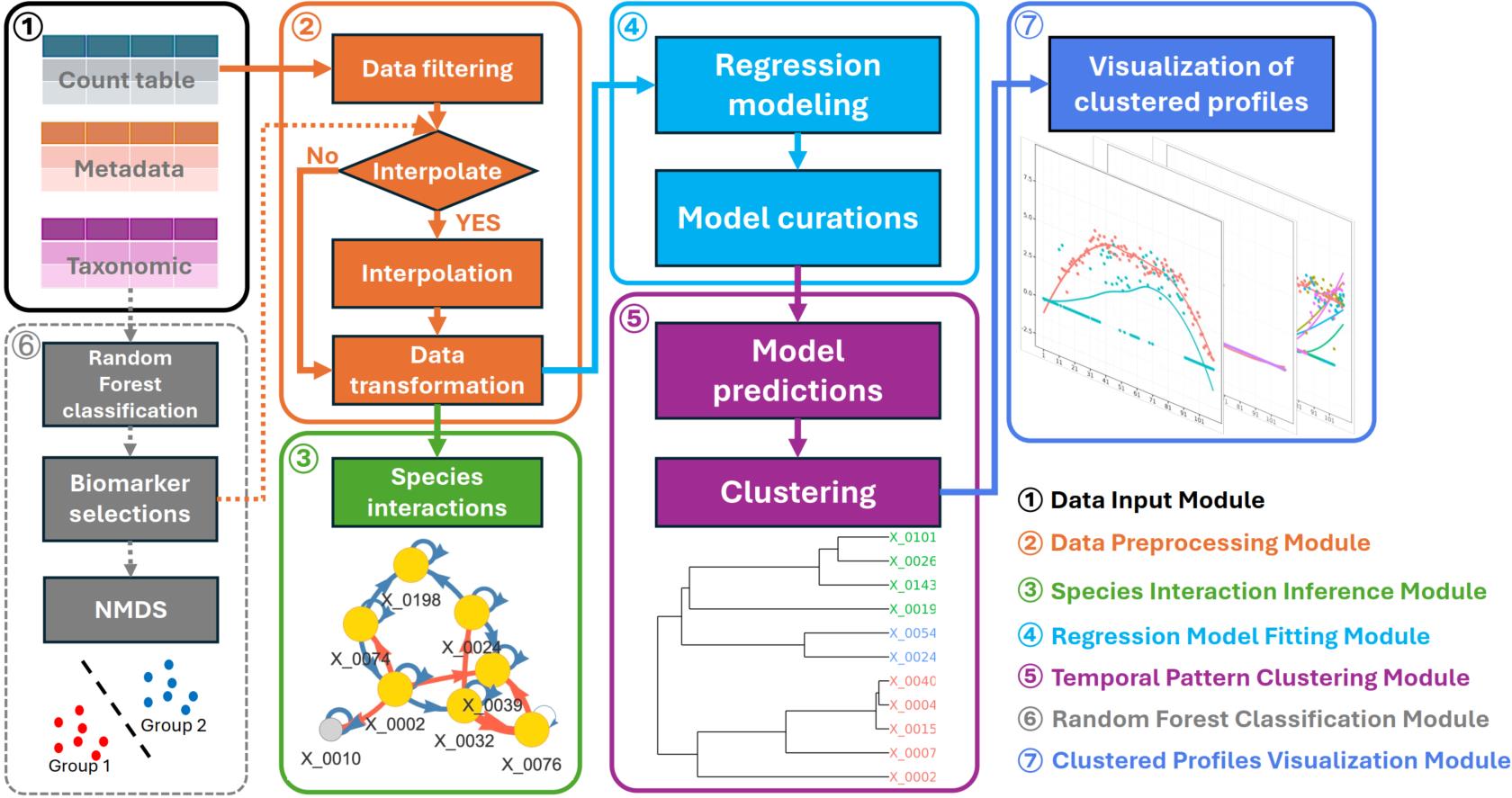
MicrobTiSDA is a user-friendly and flexible R package specifically designed for longitudinal microbiome data analysis. By integrating process-oriented functional modules—including data input, data preprocessing, interspecies interaction inference, natural spline regression modeling, temporal pattern clustering, and random forest classification—MicrobTiSDA enables users to efficiently and systematically infer reliable interspecies interactions and accurately characterize microbial abundance dynamics from time-series data. The package is compatible with diverse experimental designs and provides comprehensive, intuitive visualizations, thereby substantially enhancing both the efficiency of longitudinal microbiome data analysis and the depth of biological interpretation.
Weizmannia coagulans XY2 Mitigates Copper Neurotoxicity via Gut–Brain Axis Modulation of Tryptophan Metabolism and Oxidative-Inflammatory Crosstalk
- 18 November 2025
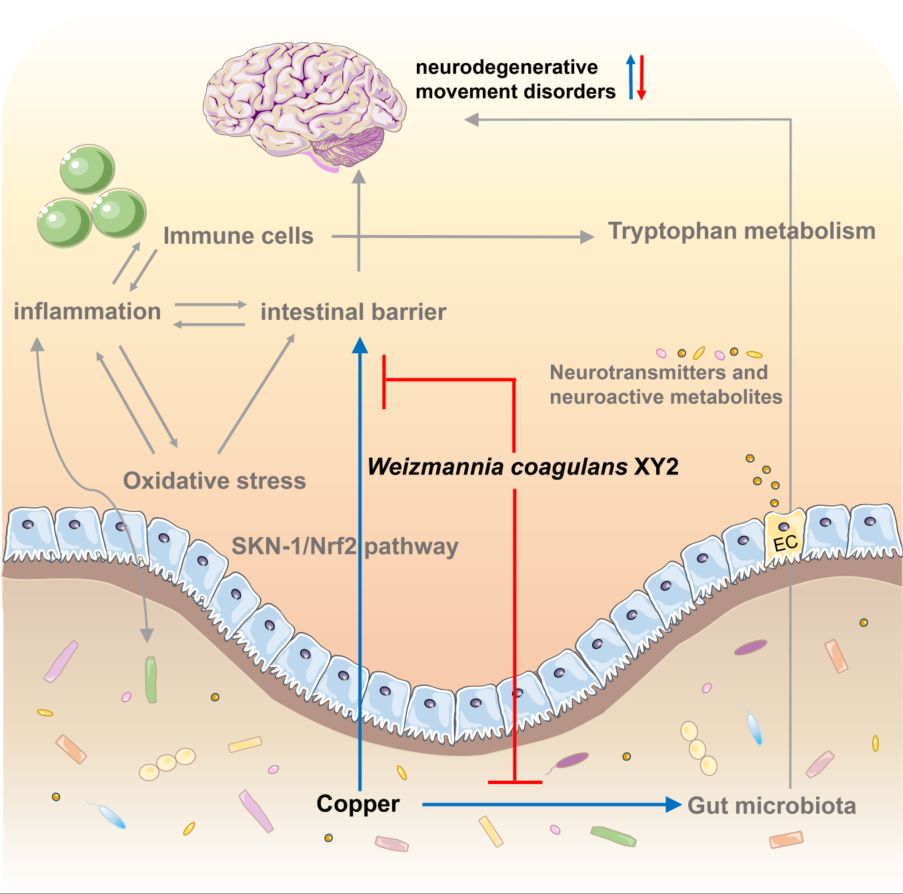
Copper interferes with tryptophan metabolism and 5-HT levels by modulating intestinal flora. Intestinal barrier breakdown and inflammatory response trigger nerve damage under copper exposure. W. coagulans XY2 alleviates copper-induced neurotoxicity by targeting a multi-dimensional “tryptophan metabolism-antioxidant defense-gut-brain axis” network.
CellOntologyMapper: Consensus mapping of cell type annotation
- 06 November 2025

CellOntologyMapper automates the standardization of cell type annotations in single-cell RNA-seq by mapping user-defined names to the Cell Ontology (CL) and Cell Taxonomy (CT). The framework constructs a comprehensive embedding database of 19,381 curated cell types using sentence transformers, large language models, and synonym expansion. User inputs, such as abbreviations (e.g., “dNK”), are processed through embedding similarity ranking and LLM amplification to resolve ambiguous or context-dependent labels. The system outputs standardized ontology terms with high accuracy (0.835 for CL, 0.911 for CT). Implemented as a Python package within the OmicVerse ecosystem and available via a web interface, CellOntologyMapper enables reproducible, large-scale single-cell meta-analysis by unifying diverse nomenclatures across studies, tissues, species, and disease states.
Impact of dietary live microbes and nondietary prebiotic/probiotic intake on osteoarthritis and rheumatoid arthritis development: Stratified findings from NHANES data
- 17 August 2024
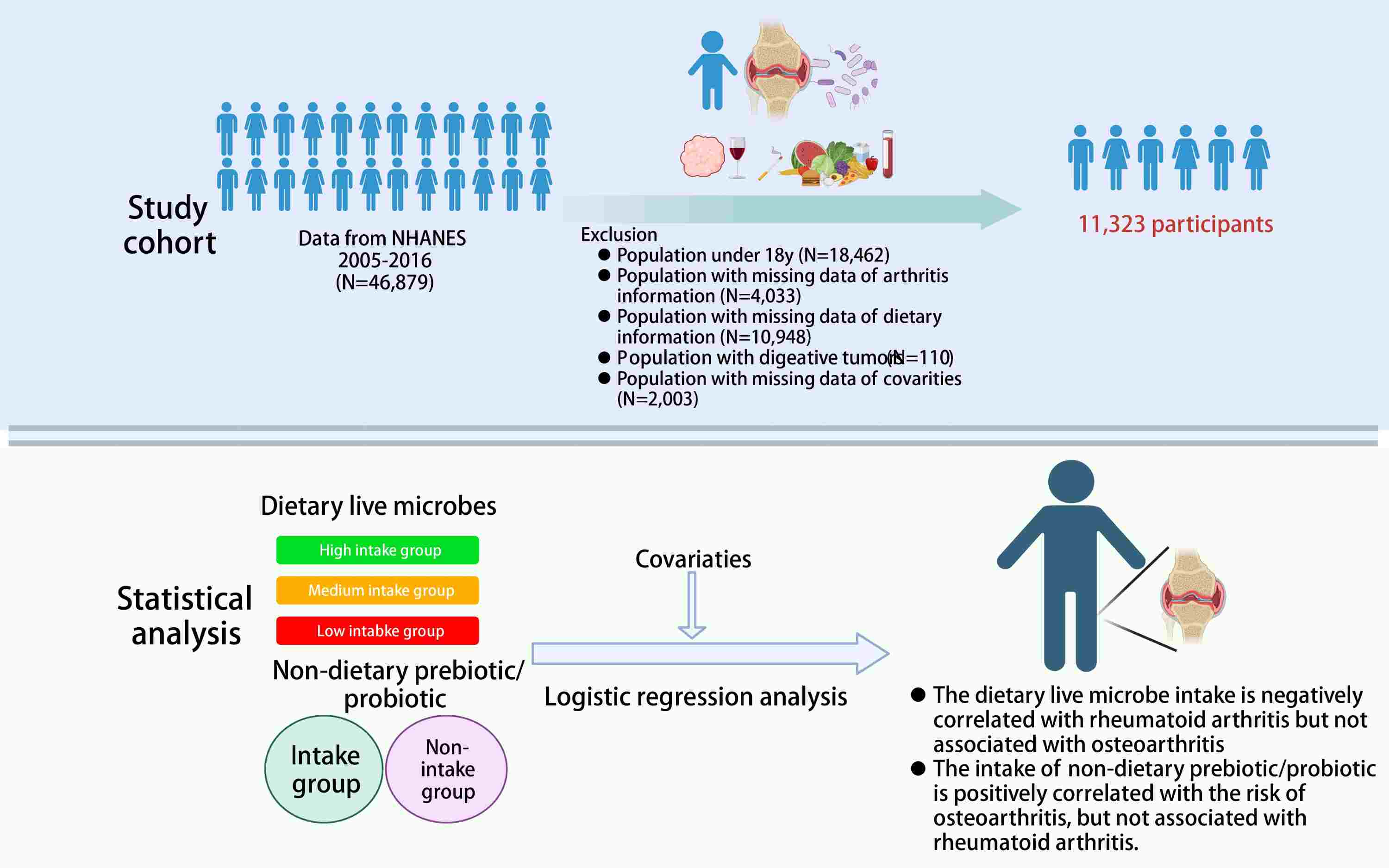
We selected participants from the NHANES database from 2005 to 2016 for this cross-sectional analysis. Logistic regression and other analytical methods were utilized to analyze the relationship between the intake of dietary live microbes and nondietary prebiotic/probiotic and the prevalence of osteoarthritis (OA) and rheumatoid arthritis (RA). The findings demonstrated a direct relationship between the consumption of nondietary prebiotic/probiotic and the prevalence of developing OA, whereas a greater consumption of dietary live microbes is associated with a lower occurrence of RA. (Graphical abstract was created with BioRender.com).
Cohort profile: A longitudinal multi-omics cohort of patients with acute coronary syndrome
- 03 July 2024
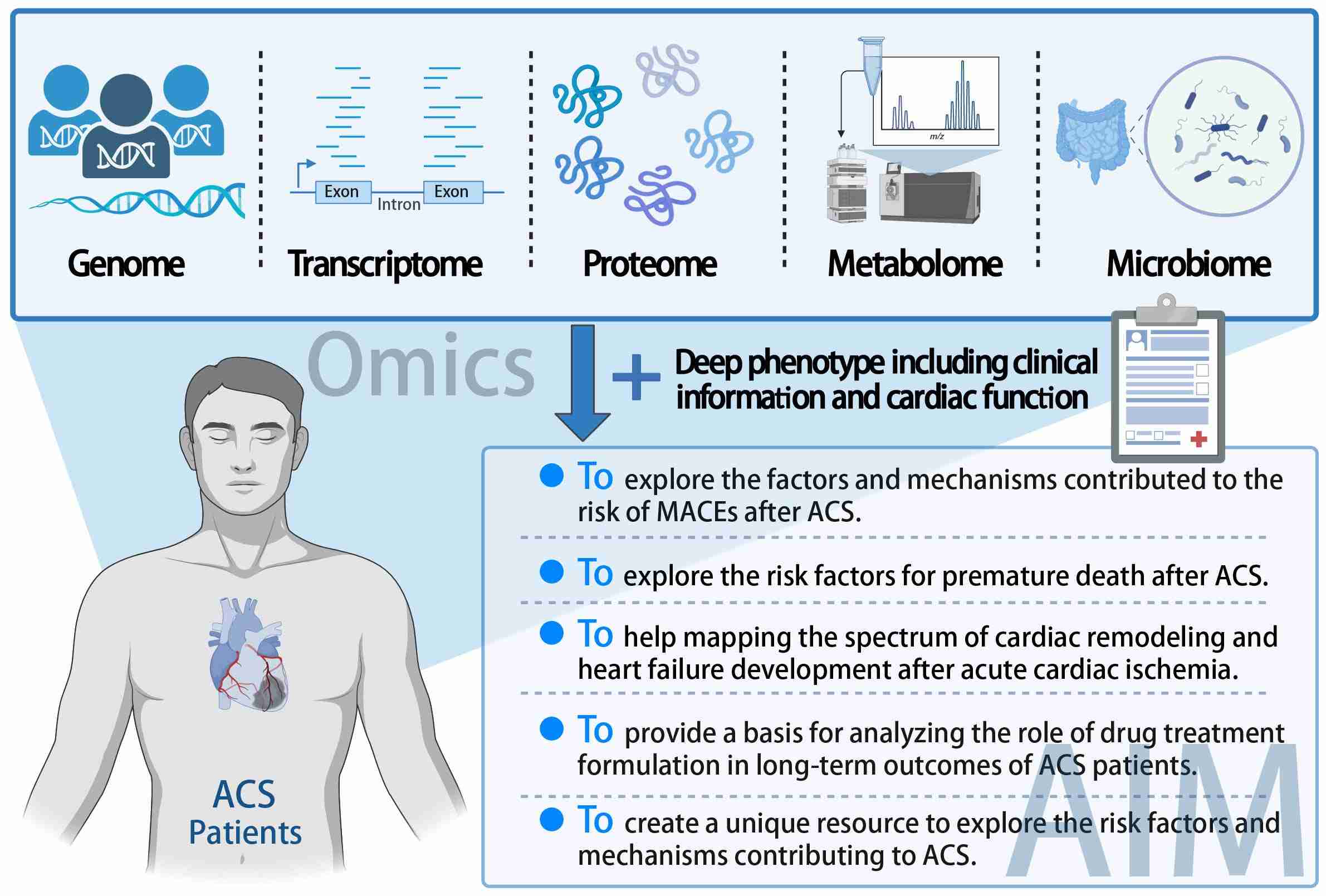
The longitudinal multi-omics cohort of patients with acute coronary syndrome (LM-ACS) is designed as a real-world prospective cohort of patients with ACS requiring coronary angiography. This study aims to enroll 50,000 participants, with a thorough collection of phenotypic data and multi-omics analyses performed on biological samples. Additionally, long-term follow-up will be conducted to track the incidence of major adverse cardiovascular events (MACEs) over the participants' lifetimes. For this purpose, three follow-up scenarios have been established for ACS patients, differentiated based on whether the patients had acute myocardial infarction (AMI) or unstable angina (UA), and whether they underwent percutaneous coronary intervention (PCI) surgery. The LM-ACS cohort seeks to create a unique resource to advance our understanding of the etiology and clinical outcomes of ACS.
The active effect of Rhizophagus irregularis inoculants on maize endophytic bacteria community
- 17 August 2024
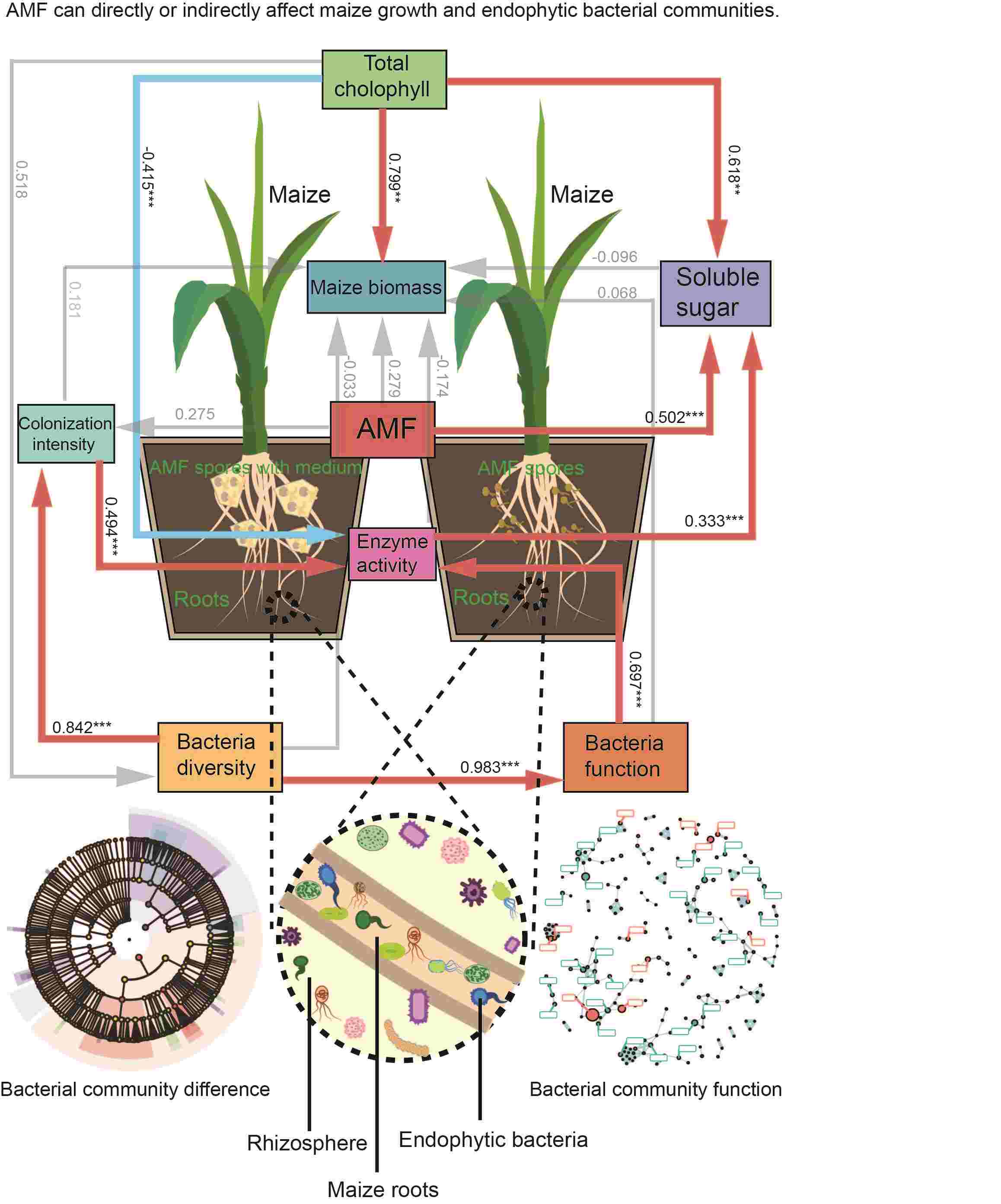
Little has been reported on the effect of arbuscular mycorrhizal fungi (AMF) inoculants from in vitro dual culture system on the growth of maize and its endophytic microbial community. Our results suggest that AMF inoculants play an important role in influencing maize growth and diversity of endophytic bacterial communities. AMF inoculants significantly promote maize growth, especially AMF inoculants with modified Strullu-Romand (MSR) medium. These inoculants also significantly increased the diversity of endophytic microbial communities, especially the abundance of beneficial bacterial flora, thus positively affecting maize growth. This study reveals the utility of AMF inoculants from in vitro dual culture system, which provides a basis for the development of environmentally friendly inoculants.
A glimpse into the future: Integrating artificial intelligence for precision HER2-positive breast cancer management
- 02 August 2024
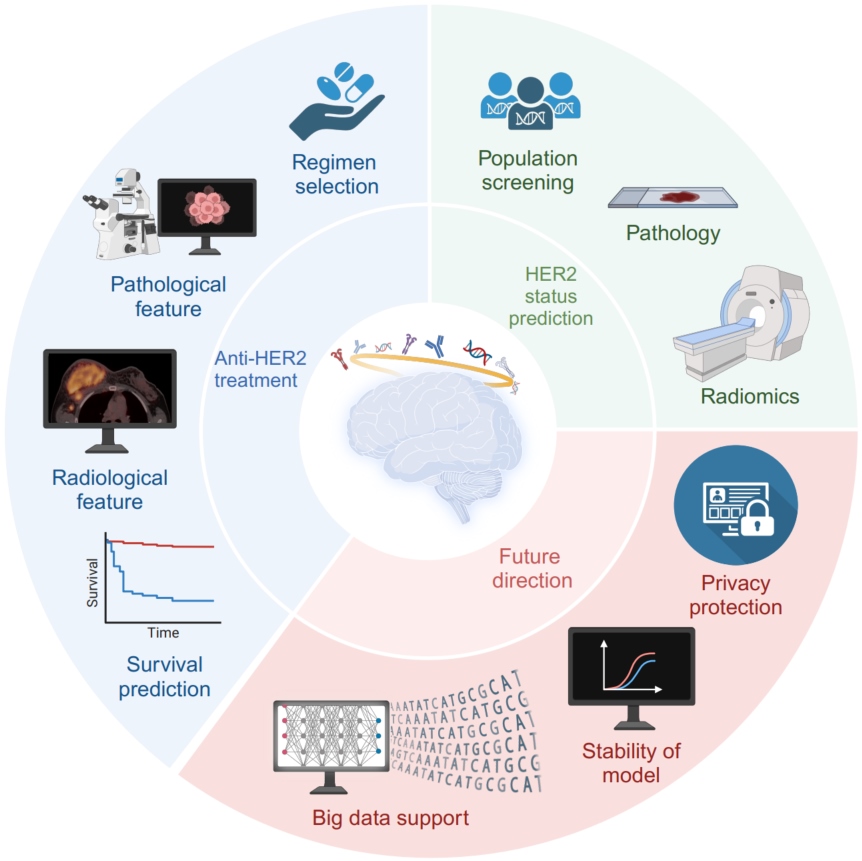
Breast cancer (BC), specifically HER2-positives subtype, has a poor prognosis. Nevertheless, the development of anti-HER2 therapy yielded satisfactory outcomes. Therefore, evaluating patient HER2 status and ascertaining responsiveness to anti-HER2 therapy is crucial. The advent of deep learning has propelled the artificial intelligence (AI) revolution, leading to an increased applicability of AI in predictive models. In the field of medicine, AI is an emerging modality that is gaining momentum for facilitating cancer diagnosis and treatment, particularly in the effective management of breast cancer. This study aims to provide a comprehensive review of current diagnostic and predictive models that utilize data obtained from histopathological slides, radiomics, and HER2 binding sites. Advancements and practical applications of these models were also evaluated. Additionally, we examined existing obstacles that AI encounters for anti-HER2 therapy. We also proposed future directions for integrating AI in assessing and managing anti-HER2 therapy. The findings of this study offer valuable insights into the evaluation of AI-based anti-HER2 therapy, emphasizing key concepts and obstacles that, if addressed, could facilitate the integration of AI-assisted anti-HER2 therapy. The integration of AI has the potential to enhance the precision and customization of screening and treatment protocols for HER2+ breast cancer.
TCfinder: Robust tumor cell discrimination in scRNA-seq based on gene pathway activity
- 07 August 2024
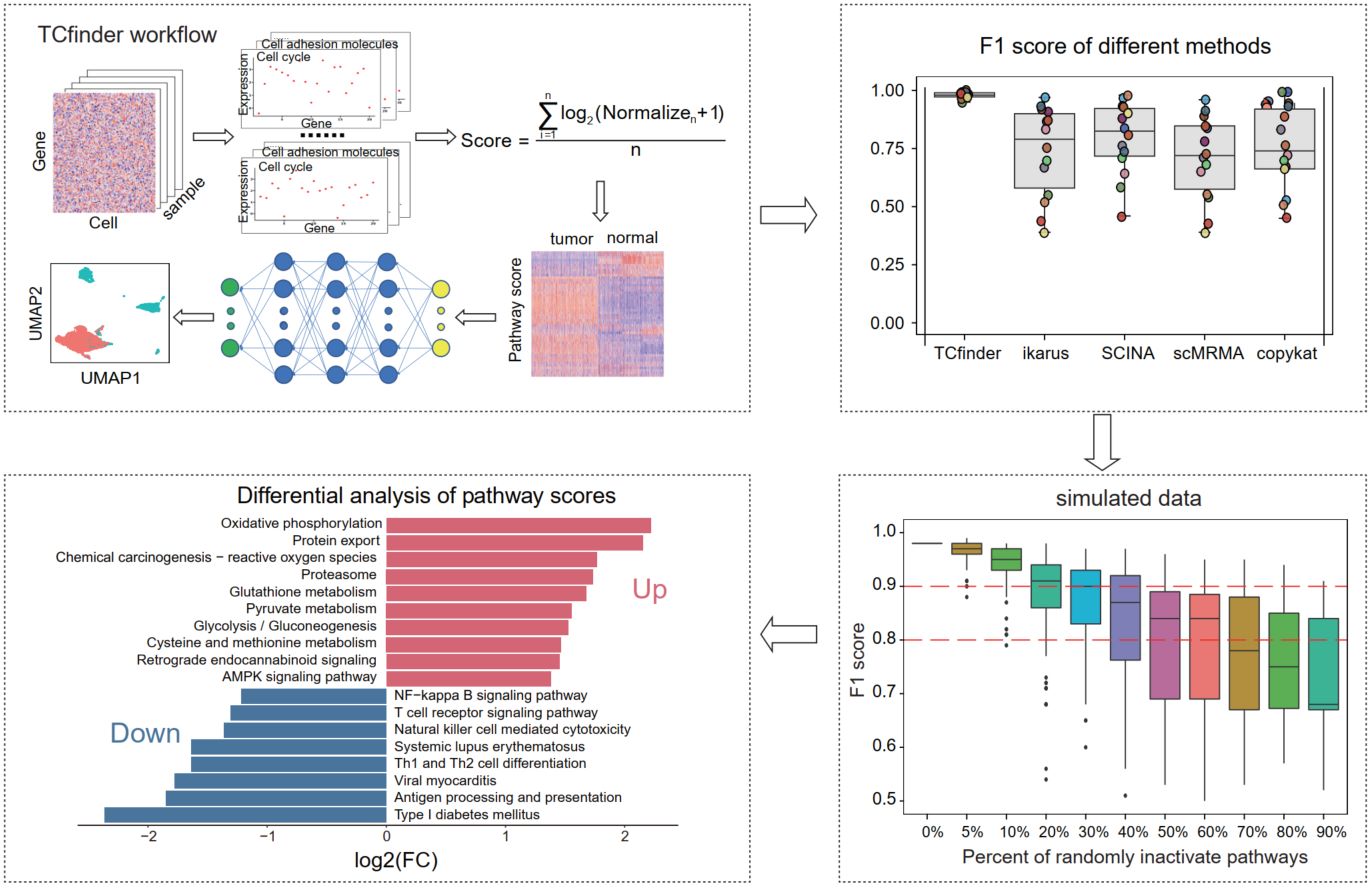
TCfinder is a tumor cell identification tool, based on pathway activity and deep neural network (DNN). Across different platforms of scRNA-seq datasets, TCfinder demonstrates robust identification efficiency. It outperforms existing tumor cell identification tools and performs under sparse data. TCfinder is freely available as an R package at: https://github.com/XSLiuLab/TCfinder.
Role of soil microbes in enhancing crop heterosis
- 01 August 2024
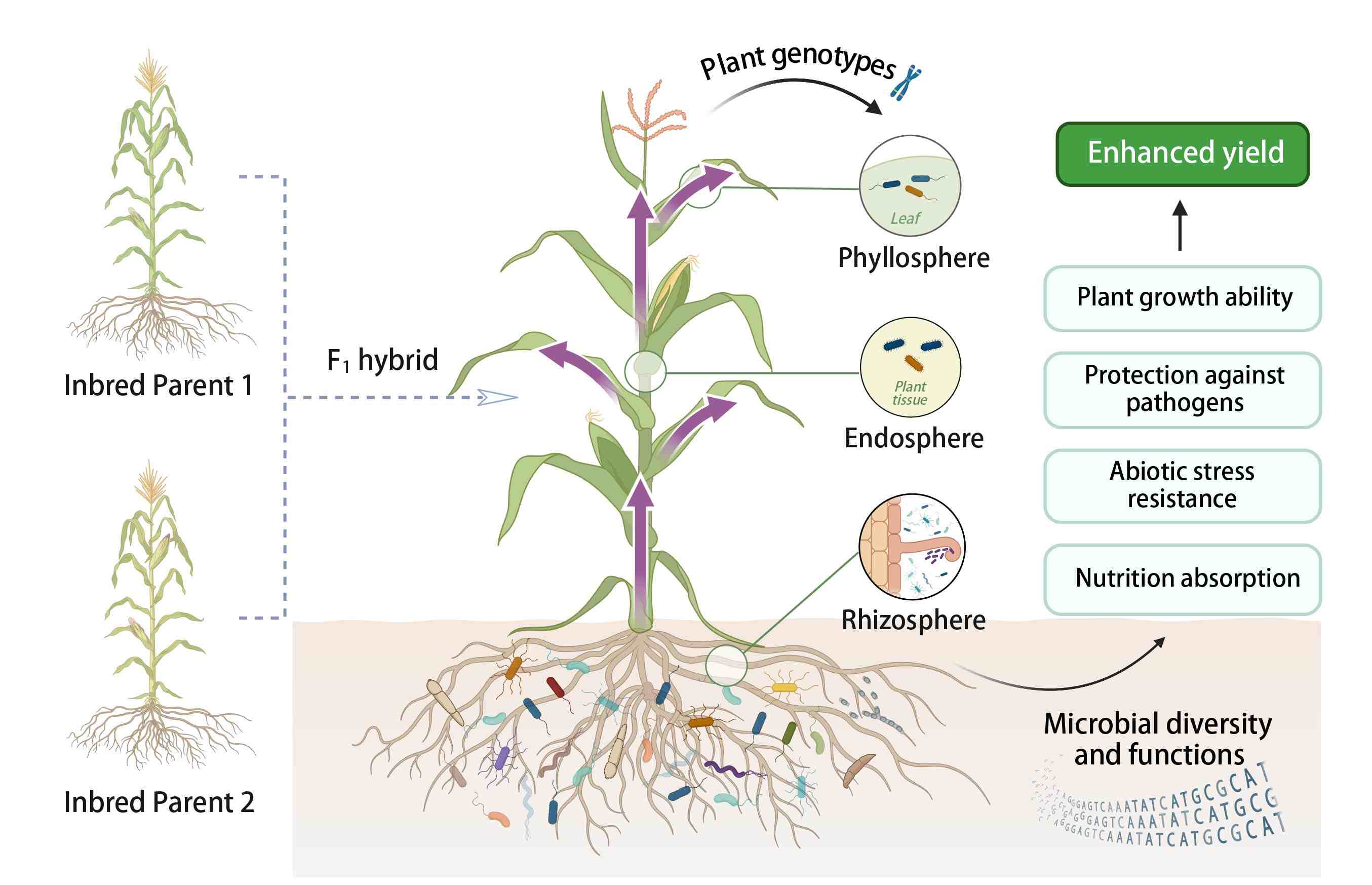
Heterosis, or hybrid vigor, is characterized by the enhanced performance of F1 progeny in terms of yield, biomass, and environmental adaptation compared to their parental lines. Recent studies underscore the significant influence of soil microbes on heterosis, revealing that plant genotypes shape microbial communities which, in turn, have the potential to support plant growth through complex host-microbe interactions. The deeper insight into microbial roles suggests innovative ways to boost crop performance and sustainability by managing the plant microbiome to further enhance heterosis
Emerging nanomedicine strategies for hepatocellular carcinoma therapy
- 04 July 2024
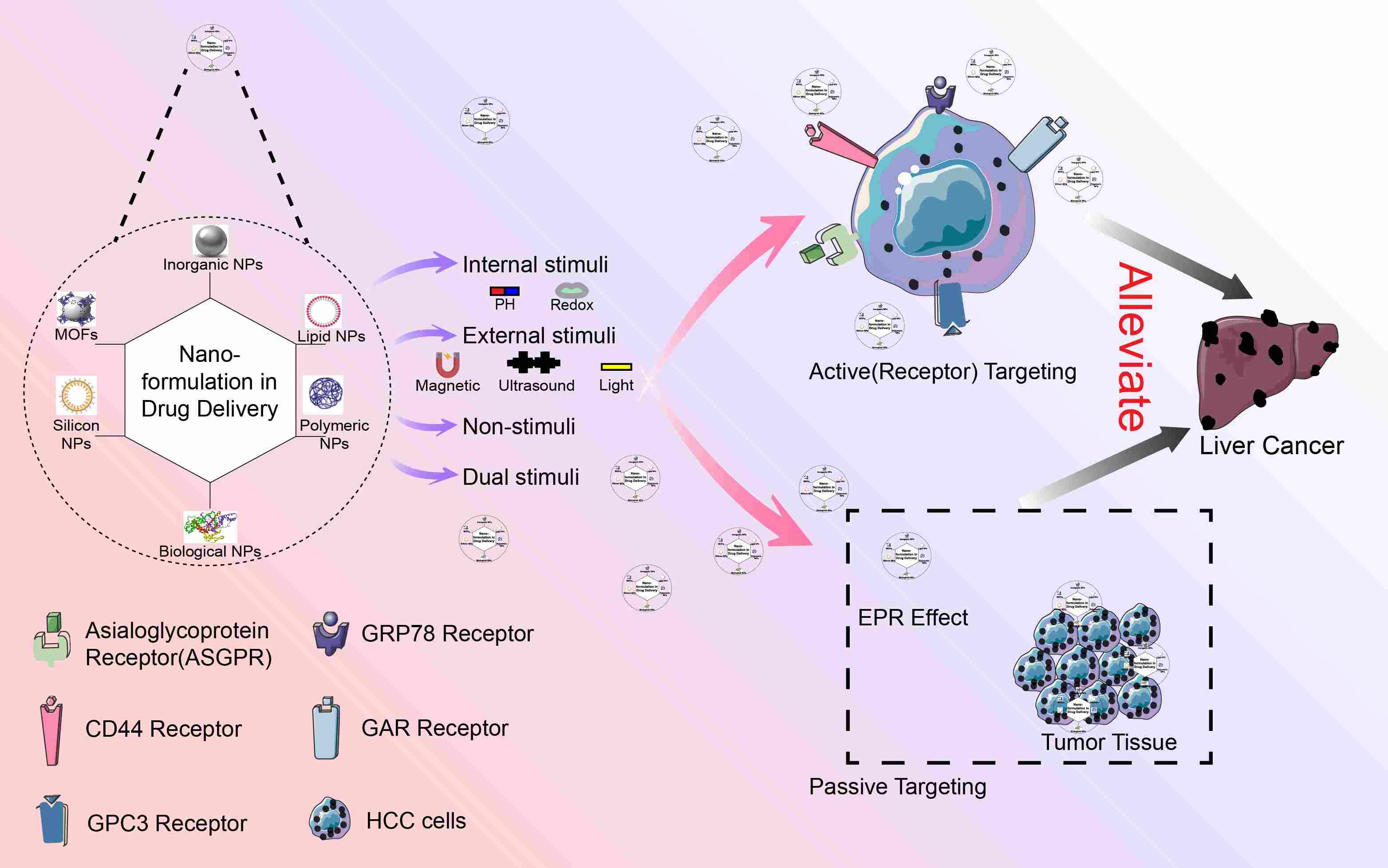
Hepatocellular carcinoma (HCC) is a prevalent malignant tumor with a range of risk factors, including viral infections, alcoholic liver disease, exposure to fungal toxins, obesity, and type 2 diabetes. Despite advancements in early detection and treatment, HCC continues to exhibit a high rate of recurrence. Patients in advanced stages have a poor prognosis, and the survival rate after cancer metastasis is notably low. Consequently, there exists an urgent necessity for novel treatment strategies. Nanomedicine possesses superior attributes, such as targeted administration and responsive drug release within the tumor microenvironment. These systems demonstrate potential in HCC treatment by facilitating the transportation of diverse therapeutic drugs. This comprehensive review aims to delve into the application of nanoparticle drug delivery systems in the management of liver tumors, with particular emphasis on formulation, targeted strategies specific to liver tumors, and various methods of drug release. By offering insights into the utilization of nanoparticle drug delivery systems in the realm of liver tumor treatment, this review endeavors to assist readers in exploring their vast potential.
Ocular microbiota types and longitudinal microbiota alterations in patients with chronic dacryocystitis with and without antibiotic pretreatment
- 09 July 2024
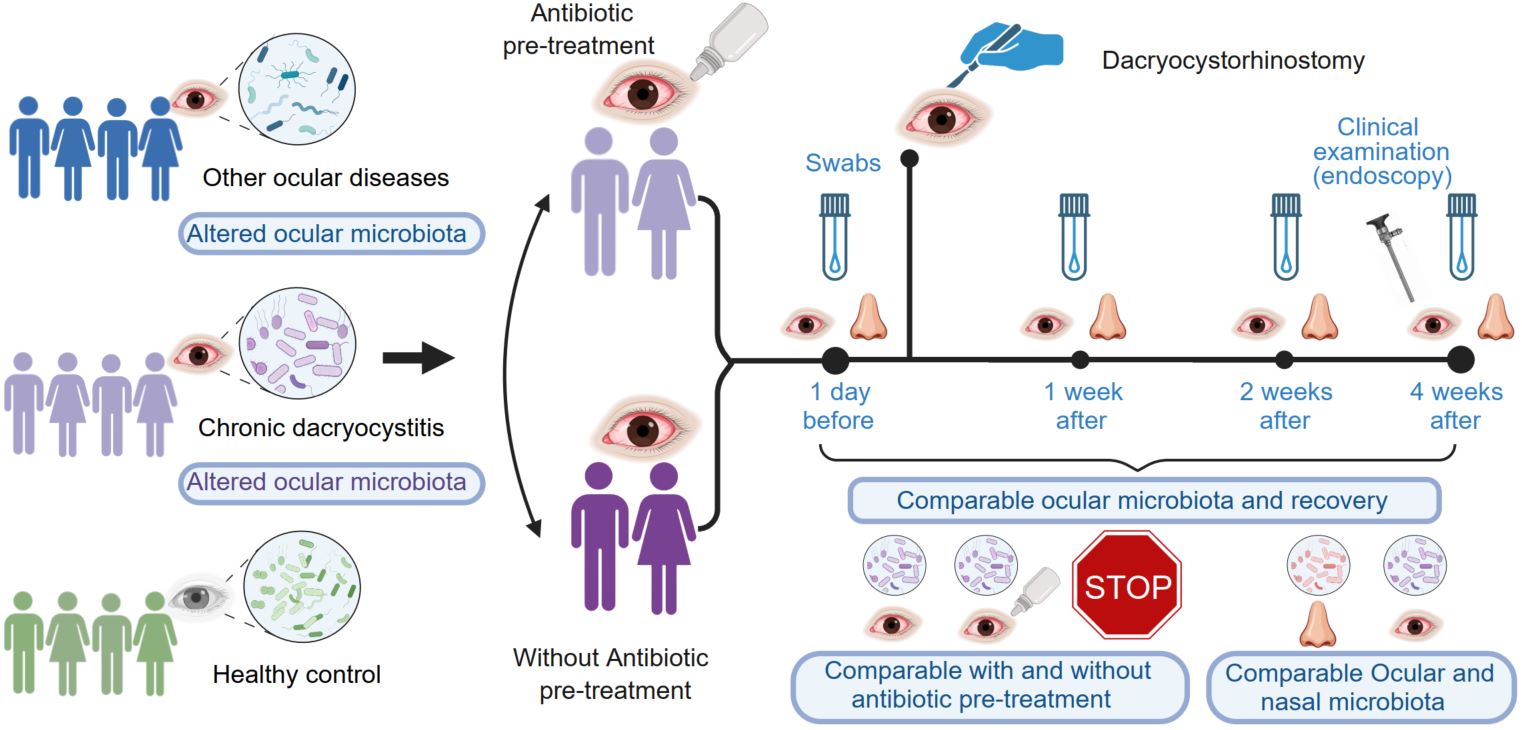
Antibiotic pretreatment is routine for chronic dacryocystitis (DC) patients. Herein, the longitudinal effects of antibiotic pretreatment before dacryocystorhinostomy for DC patients were evaluated. Conjunctival and nasal swabs were collected longitudinally from 33 DC patients with and without antibiotic pretreatment, both before dacryocystorhinostomy and at 1, 2, and 4 weeks postdacryocystorhinostomy. Additionally, conjunctival sac swabs were collected from 46 healthy volunteers and 14 other ocular diseases patients. Comparisons focused on ocular/nasal microbiota and recovery outcomes. Compared to healthy participants, DC patients without antibiotic pretreatment exhibited greater ocular microbiota diversity before dacryocystorhinostomy. Although clinical recovery rates were comparable, our results suggest that, after antibiotic pretreatment, the ocular microbiota richness and diversity, and the composition alteration tendency, significantly changed 4 weeks after surgery. This implies that the ocular microbiota was more disturbed in patients who underwent antibiotic pretreatment compared to those without such treatment. Furthermore, two types of ocular microbiota and three types of nasal microbiota were identified in ocular diseases. This study provides comprehensive data on the ocular and nasal microbiota in DC patients with and without antibiotic pretreatment, along with other ocular diseases. This finding suggested that antibiotic pretreatment may not be necessary before dacryocystorhinostomy for DC patients, especially for nonsevere cases.
Insights into the evolution and fruit color change-related genes of chromosome doubled sweet cherry from an updated complete T2T genome assembly
- 30 June 2024
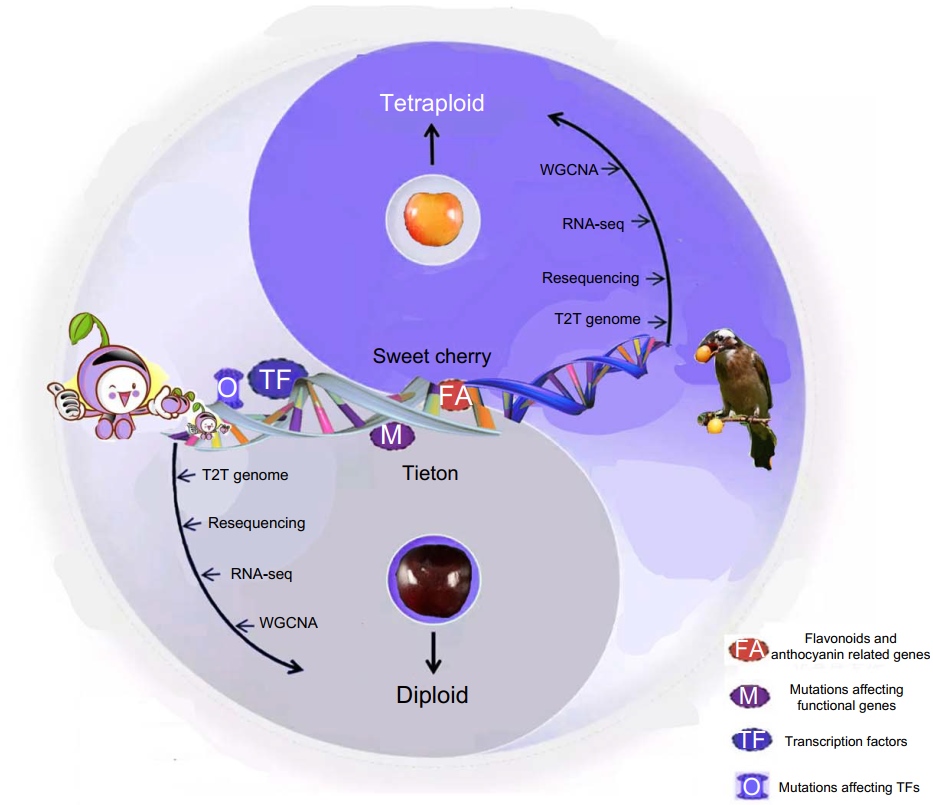
Complete T2T genome assembly of sweet cherry. Chromosome doubled sweet cherry. Mutations that affecting fruit color related genes..
Combined detection of inflammatory proteins is beneficial for diagnosing the papillary thyroid carcinoma and nodular goiter
- 02 July 2024
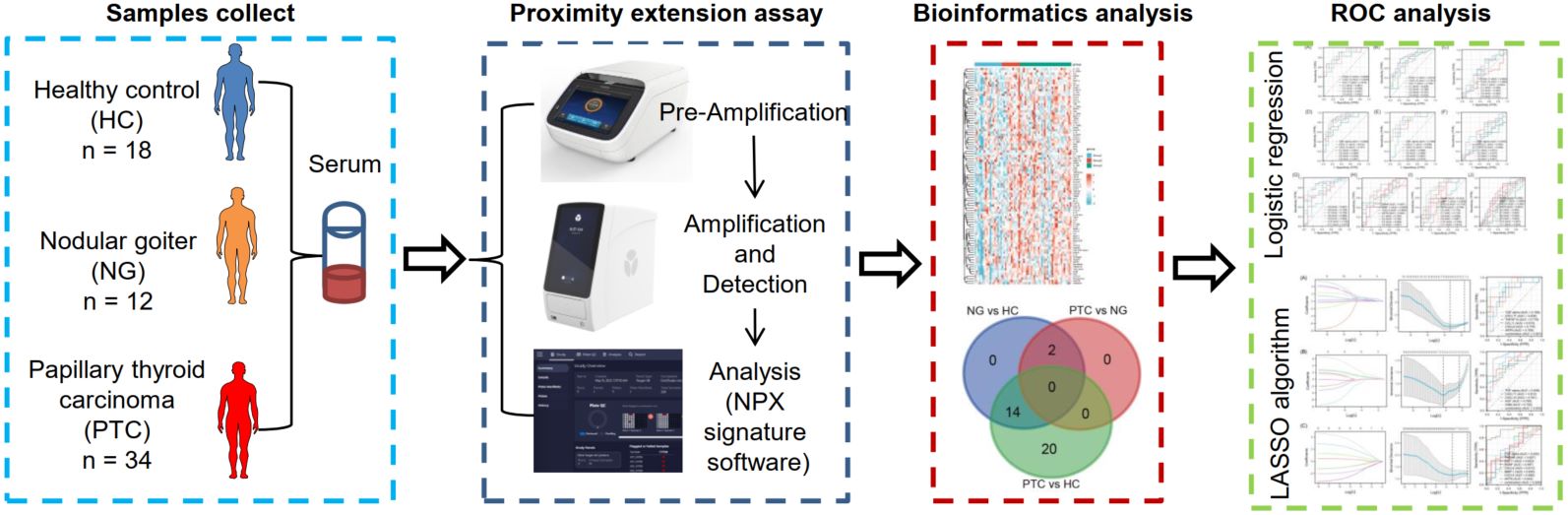
Fine-needle aspiration cytology and imaging examinations are commonly used diagnostic tools for papillary thyroid carcinoma (PTC). However, these methods have limitations. Inflammatory proteins have the potential to serve as diagnostic and prognostic markers, as well as treatment targets. The expression profile and diagnosis effect of inflammatory proteins in PTC are not well understood. Here, 18 healthy volunteers (as healthy control), 12 patients with nodular goiter, and 34 patients with PTC were collected to analyze serum inflammatory proteins by proximity extension assay. Receiver operating characteristic curve analysis was used to evaluate the diagnostic potential of differential expression of proteins via the area under the curve (AUC) analysis. A total of 36 differentially expressed inflammatory proteins were found among PTC, nodular goiter, and healthy control. The combination diagnosis derived from the logistic regression analysis exhibited promising diagnostic capabilities in distinguishing nodular goiter from healthy control (AUC = 0.88), distinguishing PTC from healthy control (AUC = 0.89), and distinguishing PTC from nodular goiter (AUC = 0.87). Whereas the combination diagnosis derived from the least absolute shrinkage and selection operator (LASSO) exhibited promising diagnostic capabilities in distinguishing nodular goiter from healthy control (AUC = 0.92), distinguishing PTC from healthy control (AUC = 0.93), and distinguishing PTC from nodular goiter (AUC = 0.93). Overall, this study offers potential biomarkers for distinguishing between PTC and nodular goiter in clinical practice. The combination derived from the LASSO algorithm outperforms logistic regression.
Developing glycoproteomics reveals the role of posttranslational glycosylation in the physiological and pathological processes of male reproduction
- 01 July 2024
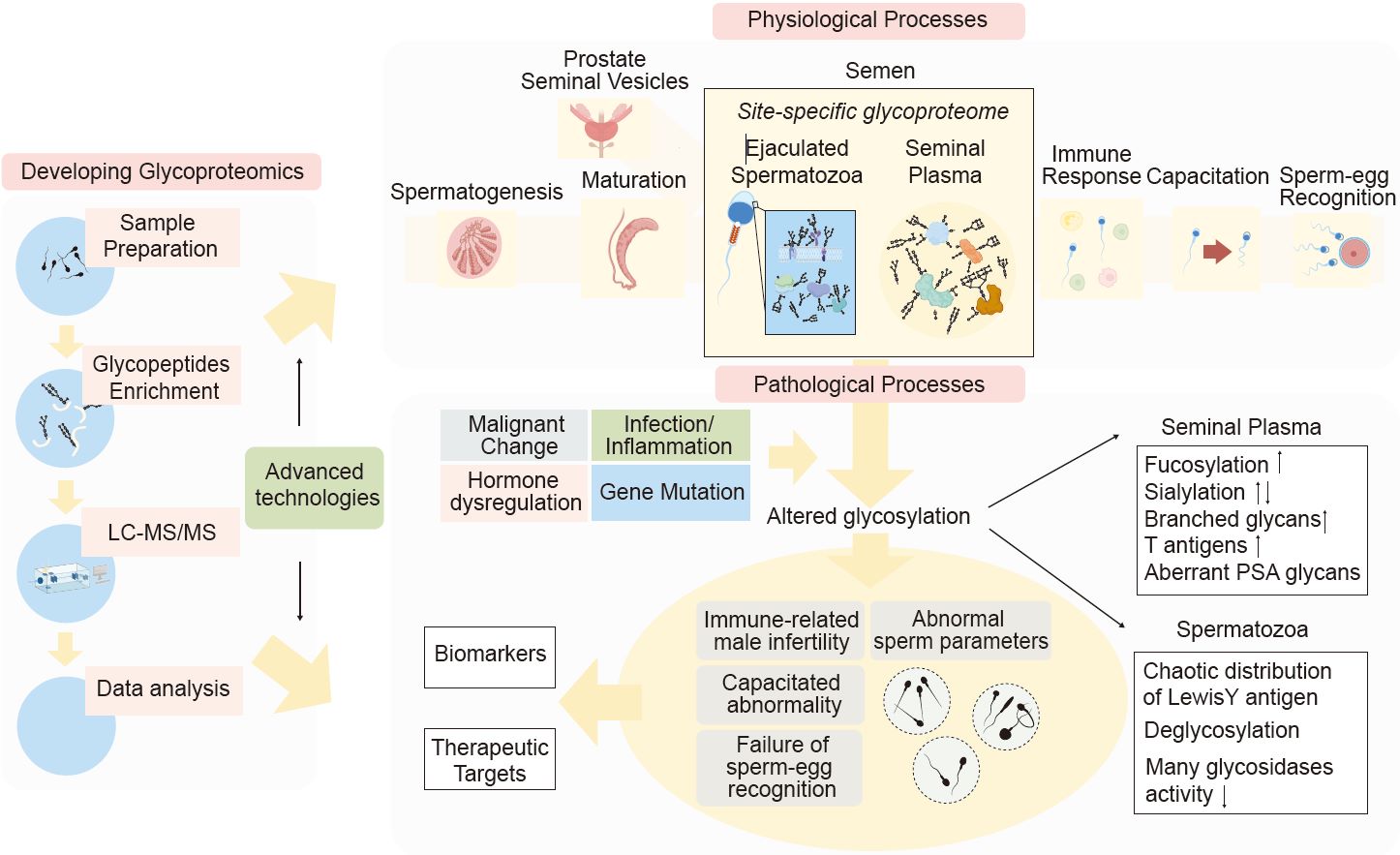
Glycosylation plays a pivotal role in the physiological and pathological processes of male reproduction. It impacts thousands of proteins and is actively ongoing through all stages of reproduction, including spermatogenesis, maturation, capacitation, and fertilization. However, our grasp on glycosylation within male reproductive processes remains limited, largely due to the technical hurdles. Recent advancements have seen the mapping of the glycoproteome of human semen, utilizing cutting-edge glycoproteomic technologies. This breakthrough lays the groundwork for in-depth research into the influence of glycosylation on male reproductive system and related disorders. Nevertheless, the field faces numerous challenges that necessitate further advancements in glycoproteomic methodologies. In this analysis, we evaluated the potential applications of advanced glycoproteomic techniques in the study of male reproduction and summarized the detailed profiling of the human semen glycome and glycoproteome. Our current understanding of glycosylation's role within the male reproductive system alongside recent progress in glycoproteomics may equip biologists with a comprehensive insight. Furthermore, this analysis brought together findings on abnormal glycosylation and its link to male reproductive disorders in the view of glycomics and glycoproteomics. It can facilitate the clinical application of glyco-related biomarkers and targets in the treatment of infertility.
A super pan-genome map provides genomic insights into evolution of diploid cotton species
- 27 June 2024
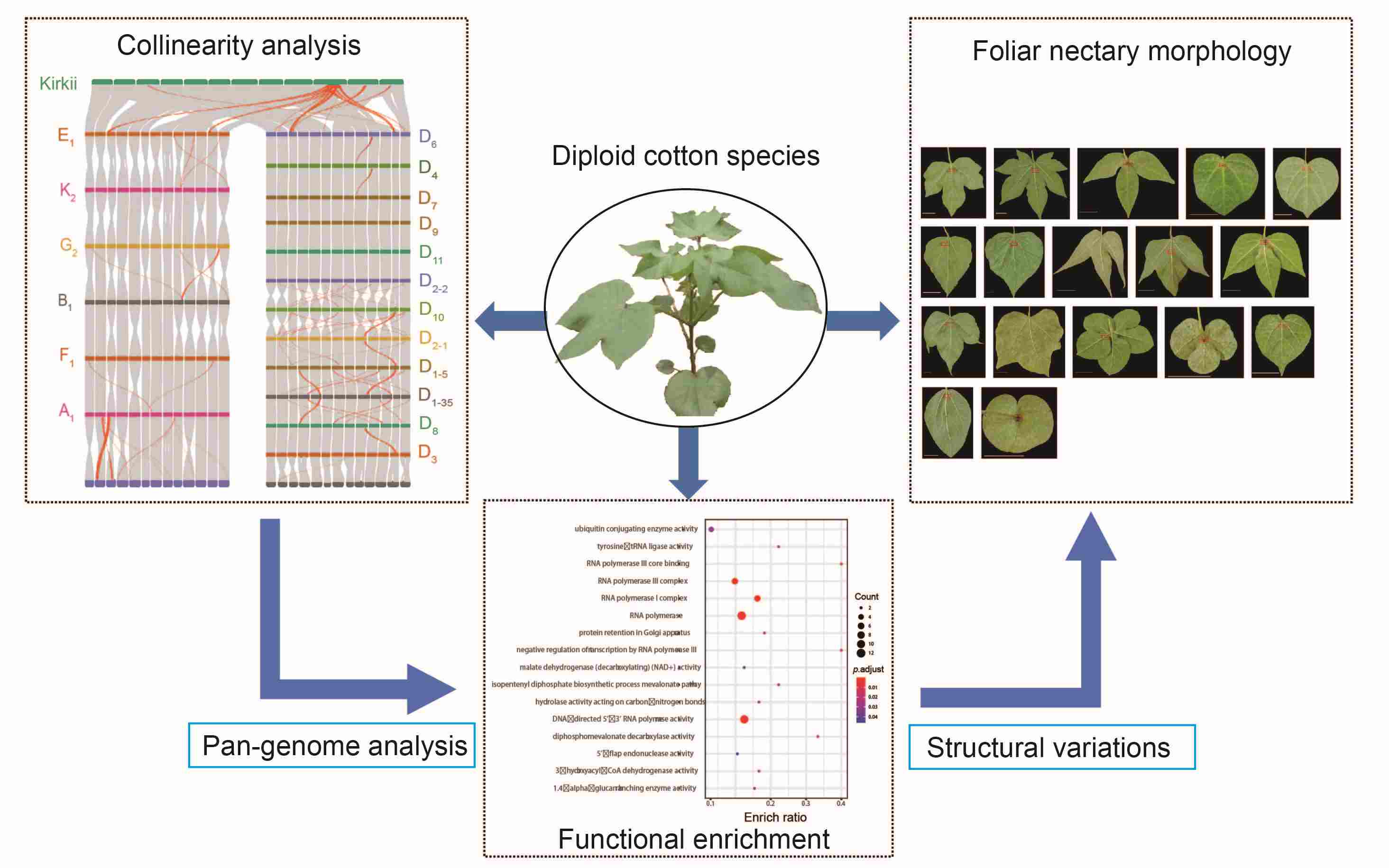
The 45 diploid cotton species identified worldwide exhibit remarkable morphological diversity. Modern cotton breeding is limited by incomplete understanding of the genetic variation in these species, suggesting a need for pan-genomic comprehensive analyses. In this study, a high-quality super pan-genome was built using 22 representative diploid cottons species and their adaptive evolution was investigated. The genomes of the twenty-two species yielded an average of 923,706 transposable elements (TEs) per assembly, with TE proportions ranging from 62.29% to 88.92%. The inferring ancestor genome structure (IAGS) showed that the D5 genome was closer to the ancestor, and the K2 genome accumulated more fissions and fusions. A gene-based super pan-genome identified 67,807 genes, including 22,384 core, 34,093 variable, and 11,330 specific genes. The structural variations (SVs) were unevenly distributed on the chromosomes, and 321 hotspot regions were detected, containing 90 genes associated with fiber initiation and/or elongation. During the eastward diffusion of diploid cotton, the genome size and structure experienced significant changes. We investigated the foliar nectary in 17 diploid cotton species, and identified a 444-bp deletion in the promoter sequence of GoNe that explained the lack of foliar nectary in G. gossypiodes (D6) and G. schwendimanii (D11). This pan-genome construction and comprehensive analysis for diploid cotton provided insight into dynamic genomic variation during diploid cotton expansion and can facilitate effective modern cotton breeding.
Long journey of 16S rRNA-amplicon sequencing toward cell-based functional bacterial microbiota characterization
- 11 July 2024
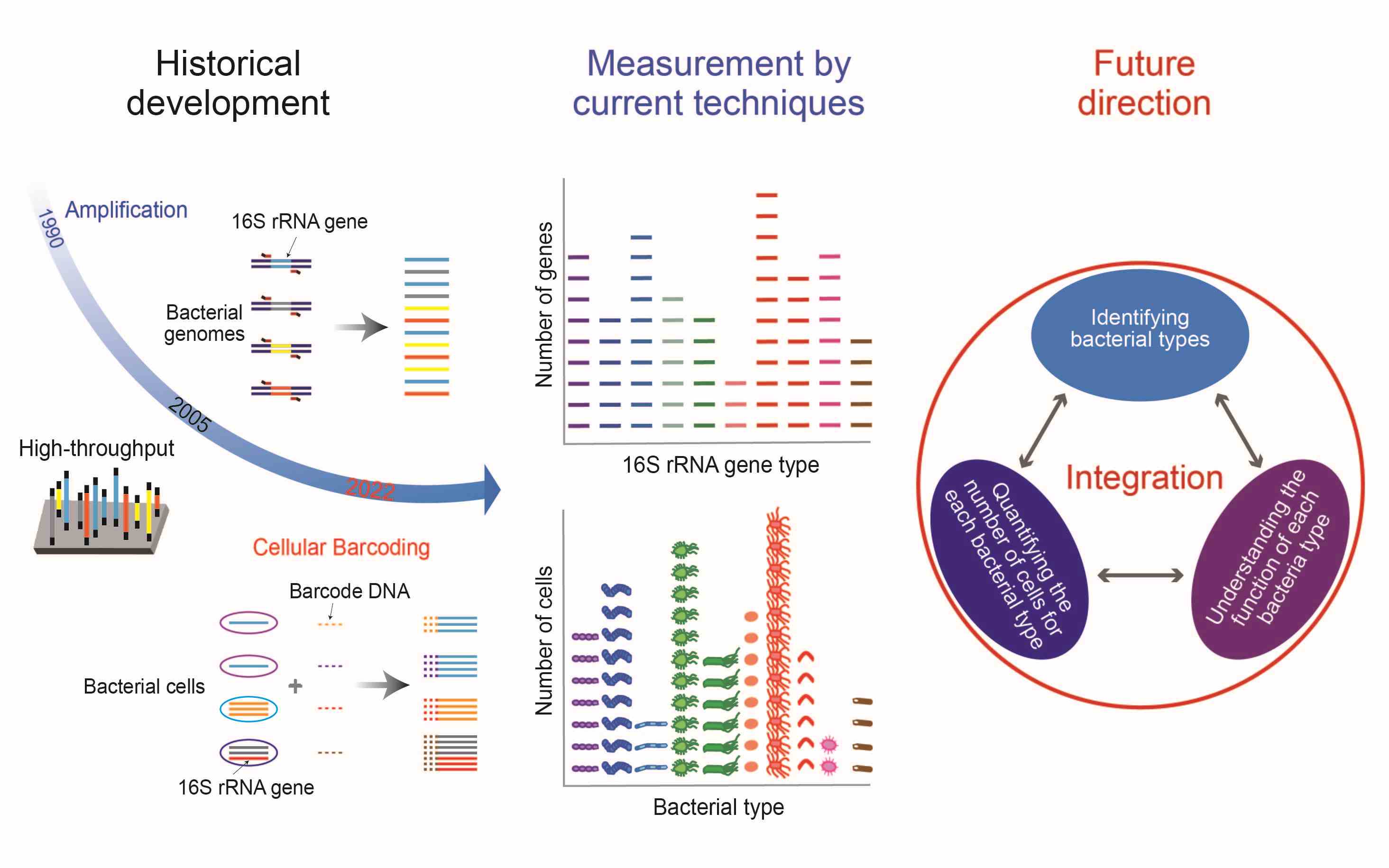
Bacteria often exist and function as a community, known as the bacterial microbiota, which consists of vast numbers of bacteria belonging to many bacterial species (taxa). Characterizing the bacterial microbiota needs high-throughput approaches that enable the identification and quantification of many bacterial cells, and such approaches have been under development for more than 30 years. In this review, we describe the history of high-throughput technologies based on 16S ribosomal RNA (rRNA) gene-amplicon sequencing for the characterization of bacterial microbiotas. Then, we summarize the features and applications of current 16S rRNA gene-amplicon sequencing approaches, including a recent achievement that enables the identification of individual cells with single-base accuracy for 16S rRNA genes and the quantification of many identified cells. Furthermore, we present the prospects for further technical development, including the combined use of high-throughput methods and other informative analyses, such as whole-genome sequencing in the common unit of the cell, which enables bacterial microbiota characterization based on both the number of cells and their functions.
STAGER checklist: Standardized testing and assessment guidelines for evaluating generative artificial intelligence reliability
- 02 July 2024
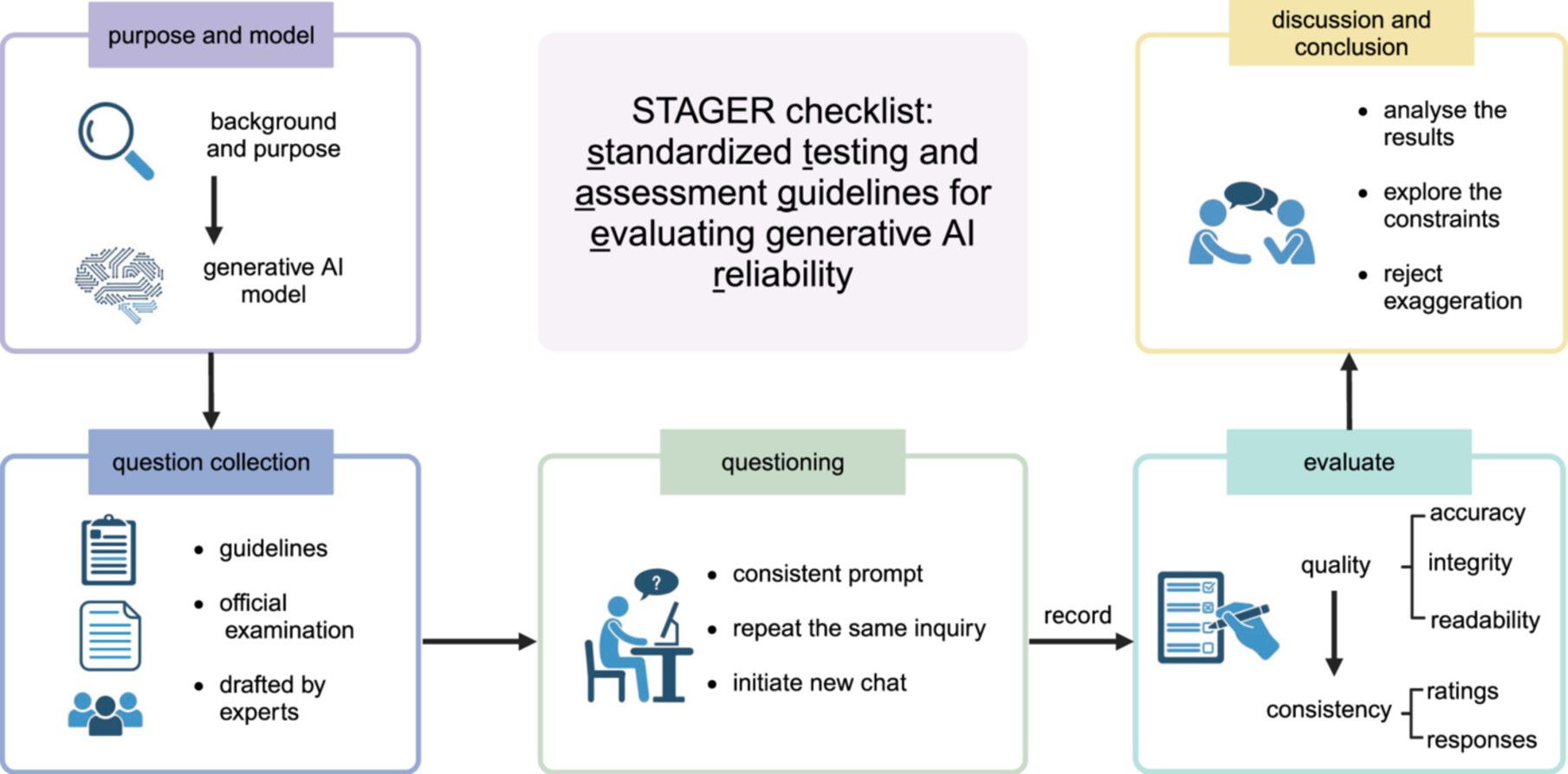
Generative artificial intelligence (AI) holds immense potential for medical applications, but the lack of a comprehensive evaluation framework and methodological deficiencies in existing studies hinder its effective implementation. Standardized assessment guidelines are crucial for ensuring reliable and consistent evaluation of generative AI in healthcare. Our objective is to develop robust, standardized guidelines tailored for evaluating generative AI performance in medical contexts. Through a rigorous literature review utilizing the Web of Sciences, Cochrane Library, PubMed, and Google Scholar, we focused on research testing generative AI capabilities in medicine. Our multidisciplinary team of experts conducted discussion sessions to develop a comprehensive 32-item checklist. This checklist encompasses critical evaluation aspects of generative AI in medical applications, addressing key dimensions such as question collection, querying methodologies, and assessment techniques. The checklist and its broader assessment framework provide a holistic evaluation of AI systems, delineating a clear pathway from question gathering to result assessment. It guides researchers through potential challenges and pitfalls, enhancing research quality and reporting and aiding the evolution of generative AI in medicine and life sciences. Our framework furnishes a standardized, systematic approach for testing generative AI's applicability in medicine. For a concise checklist, please refer to Table S or visit GenAIMed.org.
Comparative multiomics analyses reveal the breed effect on the colonic host–microbe interactions in pig
- 04 July 2024

Dysregulation of the gut microbiota often leads to immune-related disorders, indigestion, or diarrhea. Here, Jiaxing Black (JXB) pig, a local Chinese pig breed known for its great tolerance and digestibility of nutrients, was employed for a metagenomic and transcriptomic integrative analysis to reveal the gut microbiota-genes and gut microbiota-pathway interactions. A total of 452 differentially expressed genes, and 174 phyla were found between the JXB and the Duroc × Landrace × Yorkshire (DLY) pigs. Detailed analysis revealed that the differences in colon gene expression signatures between the JXB and DLY are mainly enriched in metabolic and inflammatory responses, with Lactobacillus and Lachnospiraceae enriched in DLY and JXB, respectively. Notably, Pacebacteria, Streptophyta, and Aerophobetes were found to participate in the PI3K-Akt mediated immune response in both pig breeds; however, they only accelerated the metabolism in the intestines of JXB pigs. Moreover, the host could regulate microbe metabolism and immune response by Ig-like domain-containing protein and ITIH2, PAEP, and TDRD9, respectively. Taken together, our results revealed both common and breed-specific regulations of host genes by gut microbiota in two pig breeds.









