
easynem: An R package for computing and visualizing soil nematode ecological indices
- 10 February 2026
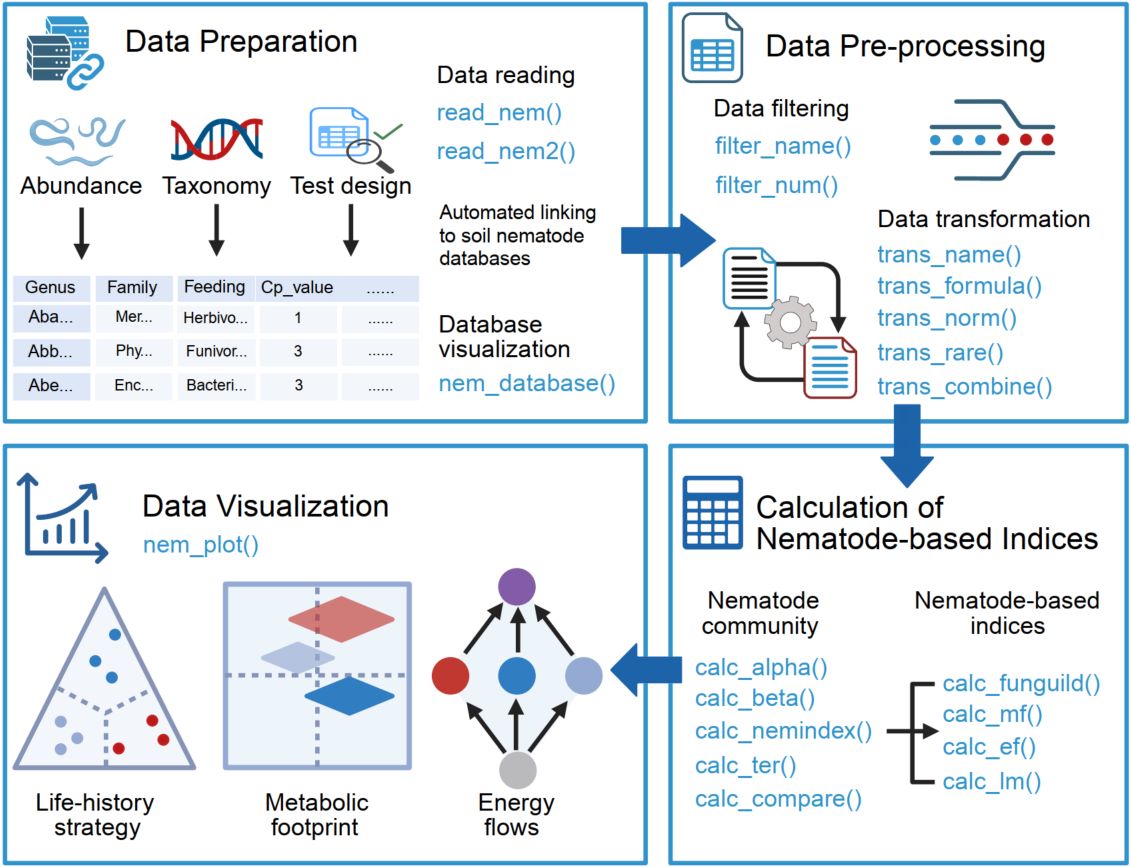
Easynem is a user-friendly and flexible R package that streamlines the complex analysis of soil nematode communities for evaluating soil health. It offers an integrated workflow for data processing, calculation of dozens of ecological indices, and comprehensive visualization of results to generate meaningful ecological insights.
Resistome flow in global landfill systems
- 06 January 2026

Antibiotic resistance genes (ARGs) represent a critical global public health threat. Effective resistome management within the One Health framework requires a comprehensive understanding of the sources and movement of ARGs across environmental media. This study provides a detailed analysis of resistome characteristics and dynamics in global landfill systems. Landfill systems were found to harbor diverse and highly mobile ARGs, with multidrug-resistant genes (MDRGs) comprising 32.63% of total ARGs, and comparable or even higher levels of associated ARG subtypes than in other environmental media, such as acidic mine wastewater. The ARG density on plasmids was 3.5 times higher than that on chromosomal sequences, with five times more abundant ARG-carrying plasmid-associated contigs in landfill systems than that in wastewater. At the metagenome-assembled genome level, members of the phylum Pseudomonadota were identified as the most abundant hosts (29.72%), harboring 47.74% and 56.47% of the total ARGs and MDRGs, respectively. Genomic analysis of 35 Pseudomonas spp. strains revealed unique Pseudomonas species in landfill systems, including P. aeruginosa. Resistome flow pathways were further mapped, including inflows into landfill (via soil, wastewater, freshwater, and the human and pig gut), movement within landfills (across landfill refuse, leachate, and airborne particles), and outflows from landfills (via landfill leachate effluent, gas emissions, and refuse evacuation). The results emphasized diverse resistome development and movement within landfill systems, ultimately contributing to the environmental spread of ARGs. Thus, our study highlights the urgent need for comprehensive policies and management strategies to mitigate resistome proliferation at every stage of municipal solid waste management.
Diversity, transfer potential, and transcriptional activity of virus-carried antibiotic resistance genes in global estuaries
- 07 January 2026
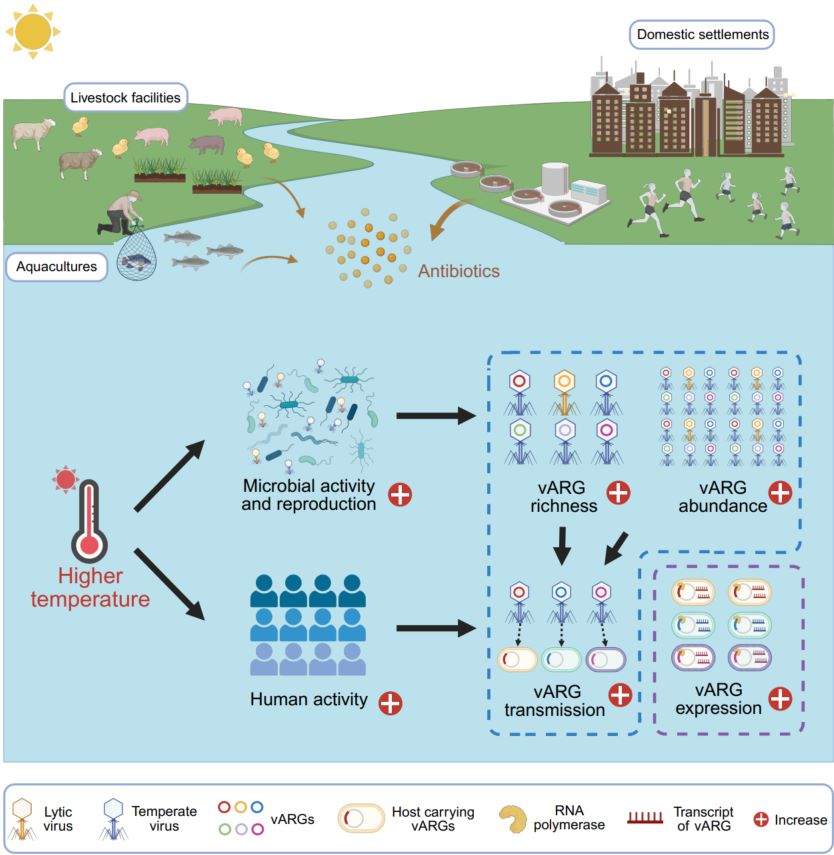
Estuaries are vital hotspots for antibiotic resistance genes (ARGs) due to substantial antibiotic pollution. Although viruses have been proposed as key reservoirs and important disseminators of ARGs in environments, their contribution to the estuarine antibiotic resistome remains largely unknown. Here, by constructing a Global Estuarine Virome dataset from 534 worldwide estuarine metagenomes, we found that estuarine viruses, particularly temperate viruses, carried abundant and diverse ARGs, and over half of the virus-carried ARGs (vARGs) were actively expressed in estuarine environments. Further analysis based on sequence similarity and minimal evolutionary distance revealed that vARG-carrying viruses targeted a wide range of prokaryotes and likely facilitated horizontal gene transfer of vARGs between viruses and prokaryotes. Importantly, elevated temperatures and coastal human activities could significantly increase the richness, abundance, transmission risk, and relative expression of estuarine vARGs. These findings provide important implications for managing the estuarine antibiotic resistome.
The evolutionary landscape and serotypic dynamics of avian infectious bronchitis virus from spike protein
- 19 January 2026
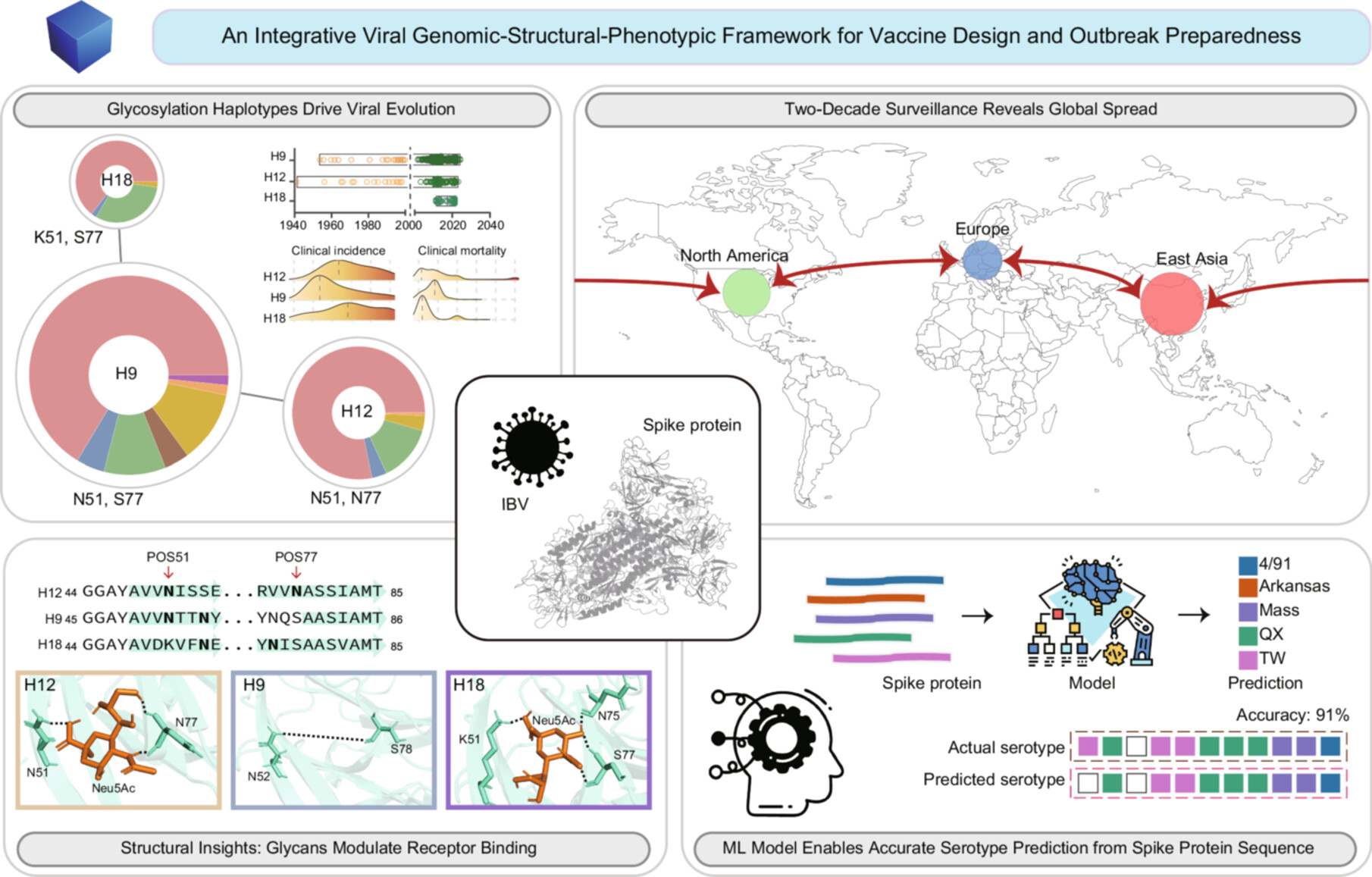
This study integrates two decades of surveillance with genomic and structural analyses to decipher how spike protein glycosylation haplotypes drive avian coronavirus evolution. We uncover how specific glycosylation patterns associate with receptor-binding affinity, shape global transmission dynamics, and correlate with clinical outcomes. Furthermore, we develop a machine learning framework for highly reliable serotype prediction directly from spike protein sequences. This multidisciplinary work establishes a scalable “viral genomic-structural-phenotypic” framework to forecast coronavirus evolution and guide vaccine design.
Oleoylethanolamide regulates intestinal stem cell activity and villus size via PPARα signaling pathway
- 26 January 2026
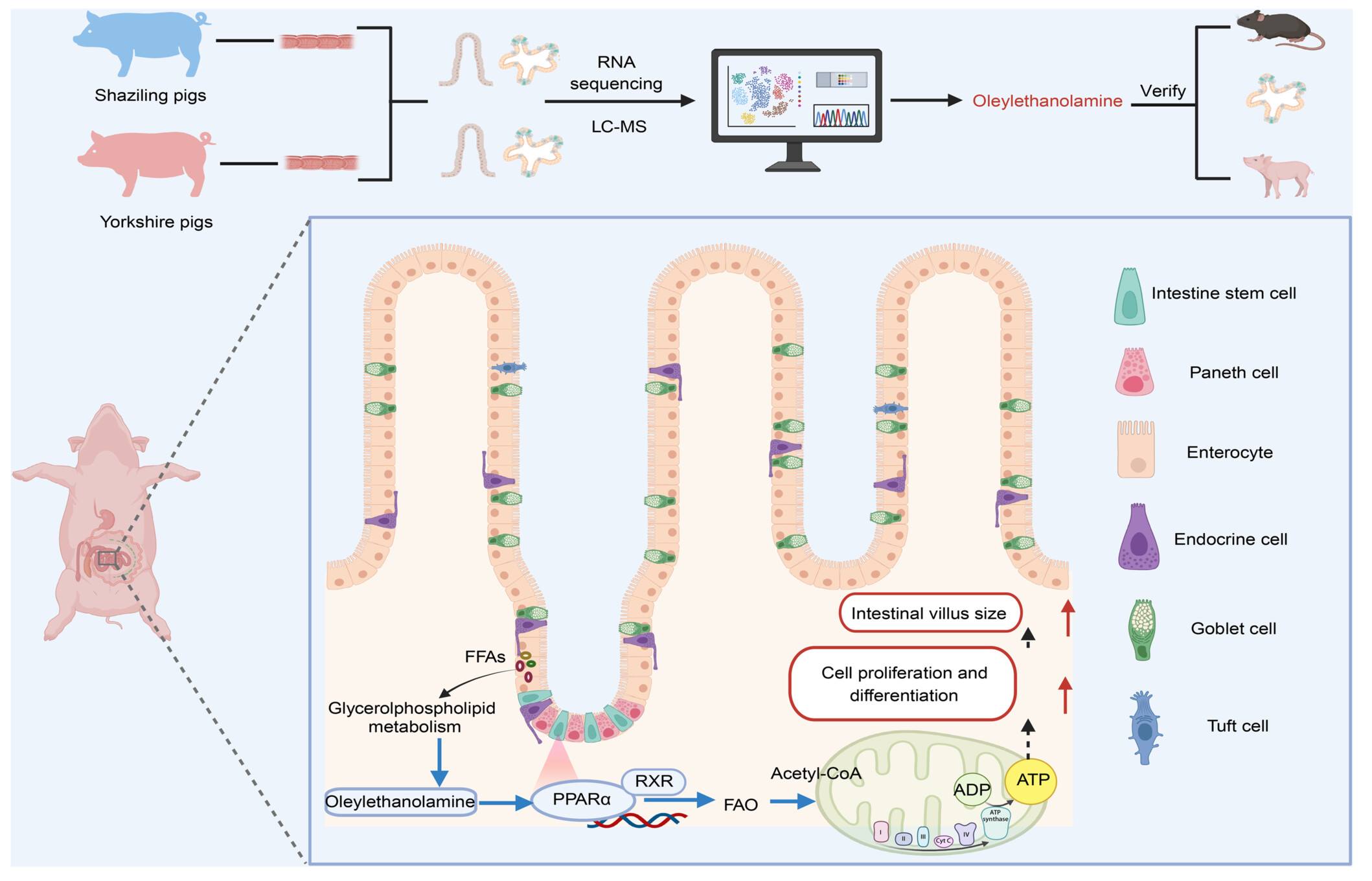
First use of a natural swine model with lipid metabolism to directly link lipid differences to a quantifiable intestinal villus height phenotype. Identified the phospholipid-derived oleoylethanolamide, rather than canonical fatty acids, as the endogenous primary ligand that activates peroxisome proliferator-activated receptors α (PPARα) to enlarge villi. Established and validated a PPARα-oxidative phosphorylation–stem cell energy switch axis, offering a druggable metabolic node that couples nutrient status to morphogenesis.
ECCFP: A consecutive full pass-based bioinformatic analysis for eccDNA identification from long-read sequencing data
- 03 February 2026
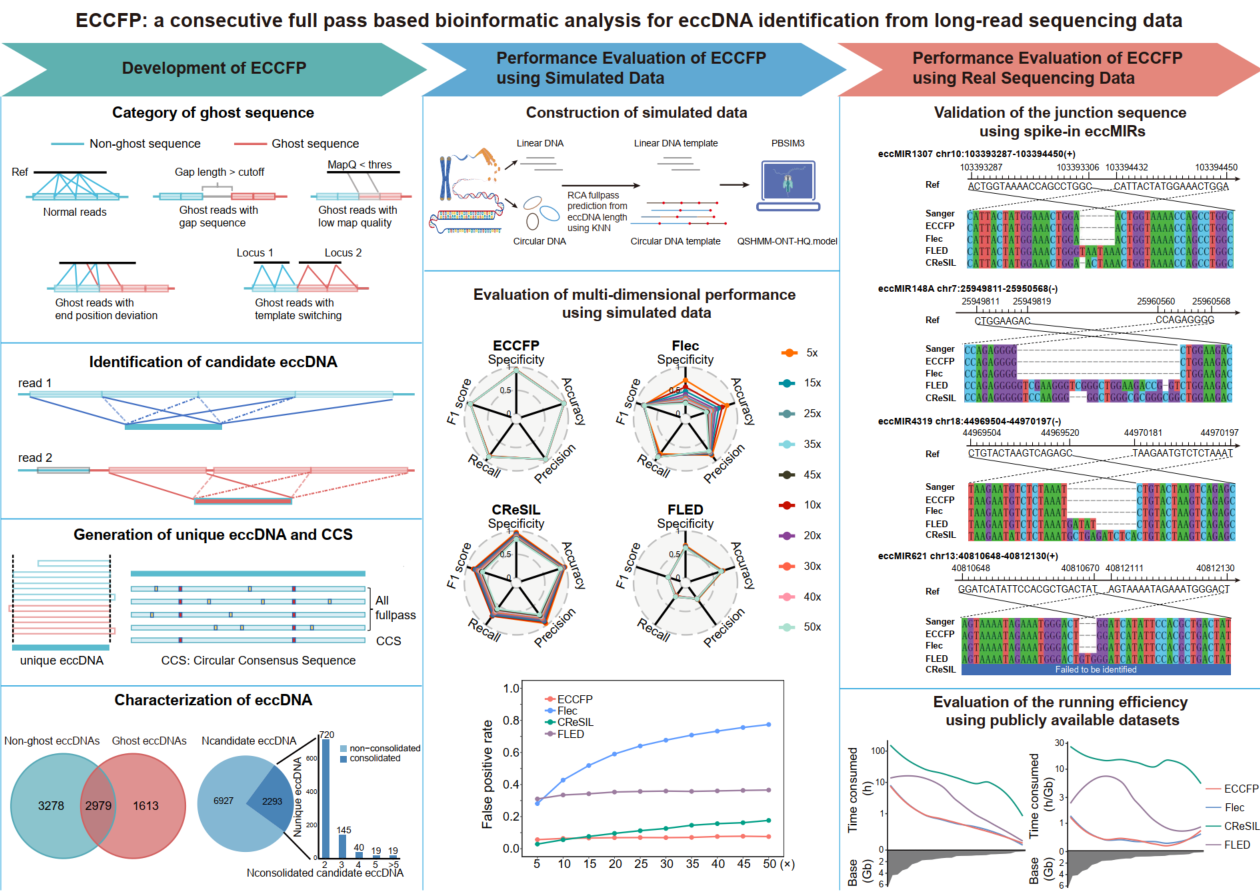
A new bioinformatics pipeline called ECCFP has been developed to improve the detection of extrachromosomal circular DNA (eccDNA) from long-read sequencing data. ECCFP uses all consecutive full passes from individual reads for candidate eccDNA identification and consolidates candidate eccDNAs to generate accurate unique eccDNA. To thoroughly evaluate the performance of ECCFP, 6 simulated datasets, 2 real sequencing datasets with spiked-in artificial synthesized eccDNAs and 29 publicly available real sequencing datasets, were applied. The results demonstrate that ECCFP markedly reduces the false positive rate, significantly increases the number of unique eccDNAs detection, achieves higher accuracy in identifying junction positions and circular consensus sequences, and also shortens the runtime, when compared to other three eccDNA analysis pipelines. Overall, ECCFP provides a more efficient and accurate bioinformatic analysis for eccDNA detection from long-read sequencing data, which might push the research forward on eccDNA as a biomarker in clinical settings.
Biosynthesis gene cluster-containing species drive microbial community stability under thermal stress through enhanced ecological cohesion
- 15 February 2026
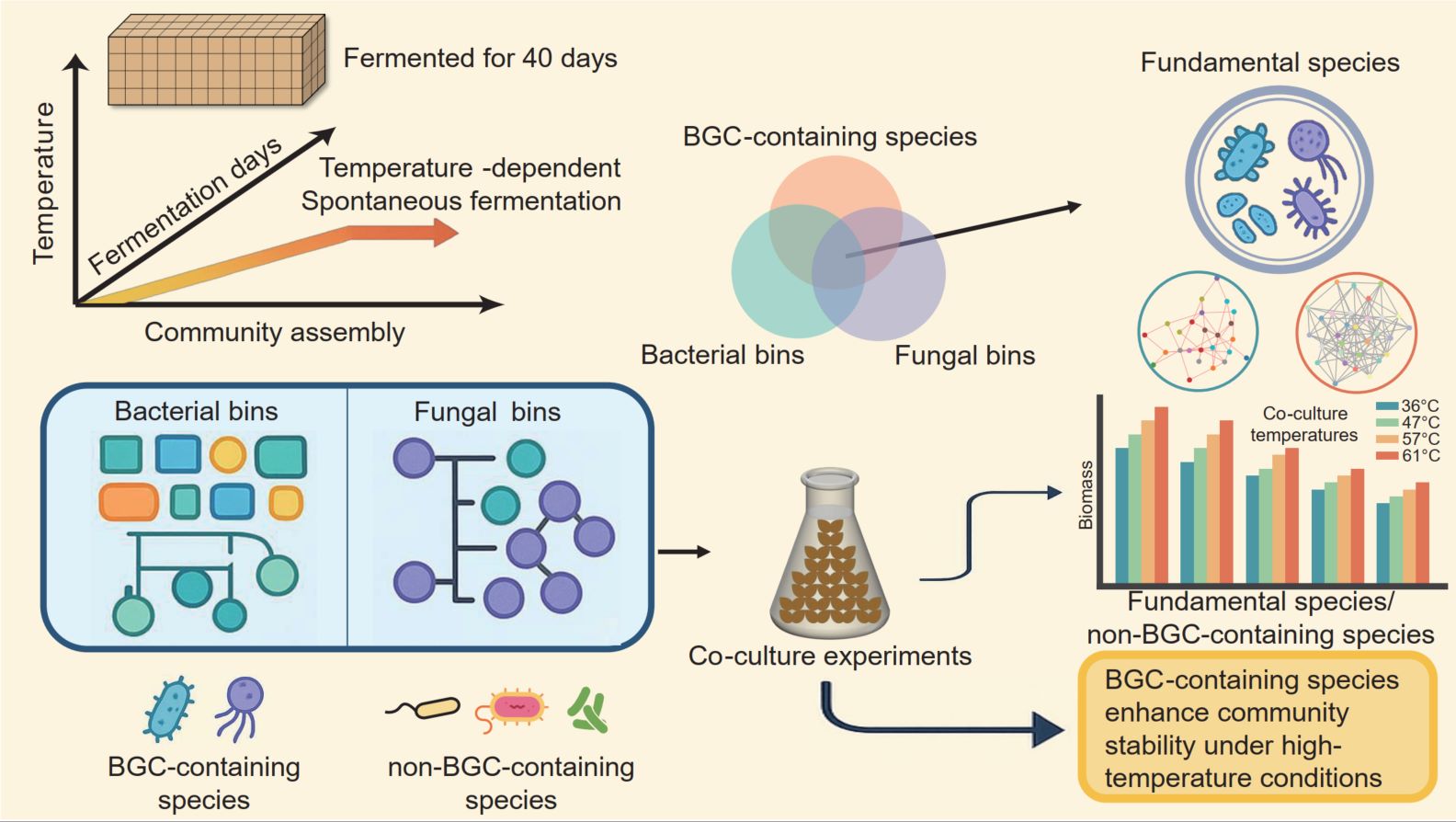
The study framework for exploring microbial community assembly and community stability of biosynthesis gene cluster (BGC)-containing species under thermal stress (20°C–65°C) during 39 days of spontaneous fermentation. Temperature gradients during spontaneous fermentation reshape microbial community assembly, driving pronounced succession in both bacterial and fungal communities. Community assembly is jointly governed by deterministic and stochastic processes, with bacterial heterogeneity selection and dispersal limitation showing strong associations with thermal stress, while fungal assembly is primarily linked to homogeneity selection and drift. In parallel, BGC-containing species are enriched at high temperatures and are associated with higher community cohesion and stability-related network properties under thermal stress. Removal simulations identified key fundamental species, indicating their importance for community resilience, while co-culture experiments demonstrated that BGC-containing species significantly increased biomass under thermal stress. These results underscore the critical role of BGC species in promoting stability and resilience.
Comprehensive assessment of unfolded protein response and its association with tumor progression in pan-cancer
- 20 February 2026
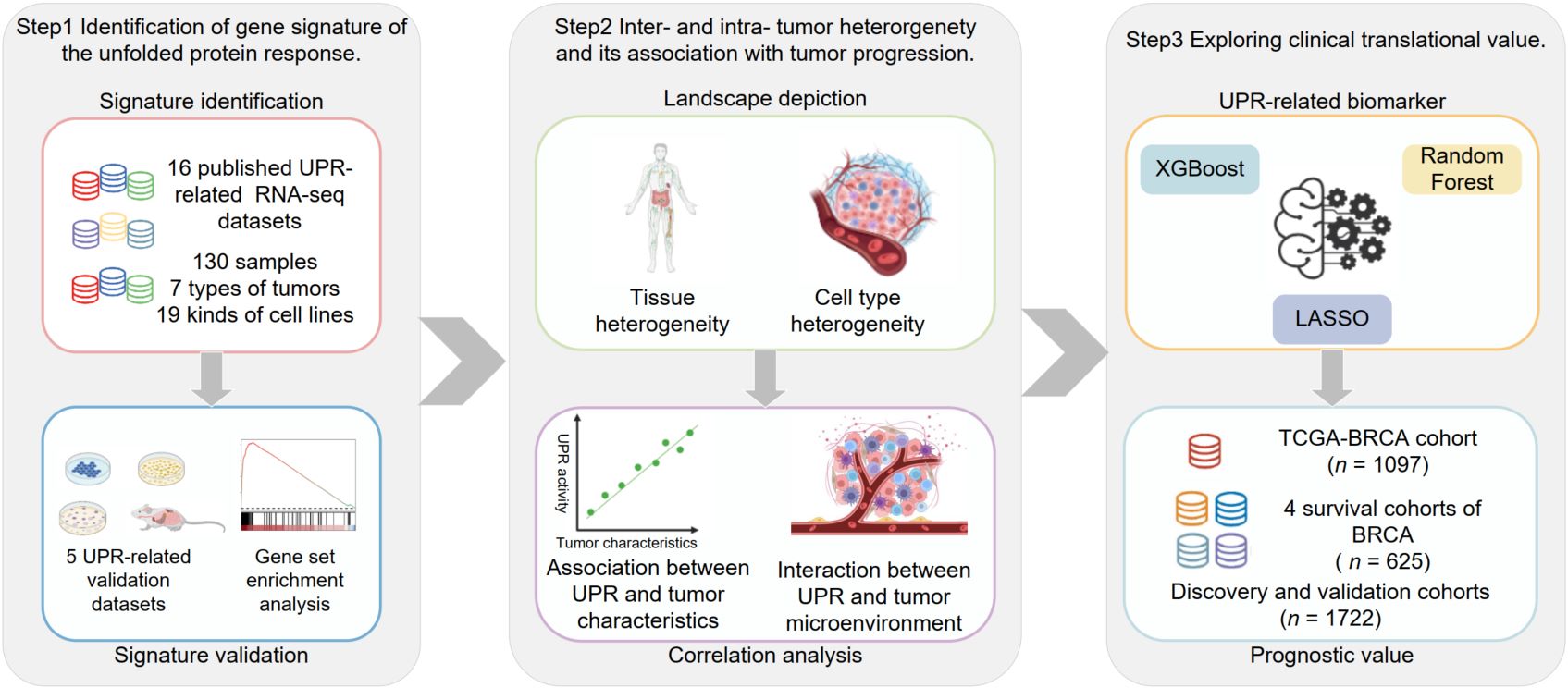
Schematic of the study design. In this study, we developed a comprehensive unfolded protein response (UPR) gene signature and applied it to systematically characterize the UPR landscape across multiple cancer types. By integrating bulk and single-cell transcriptomic data, we revealed substantial inter- and intra-tumoral heterogeneity in UPR activity and uncovered critical associations between UPR and key cancer features, including genomic features and immune responses. Importantly, we further employed machine learning methods to construct a simple three-gene prognostic model derived from the UPR signature, which can effectively predict overall survival of breast cancer patients, highlighting both the role of UPR in modulating tumor immunity and the clinical utility of the UPR gene signature in cancer prognosis and precision medicine.
Intact pulse cotyledon cell diet improves systemic growth via the somatotropic axis in malnourished mice
- 25 February 2026
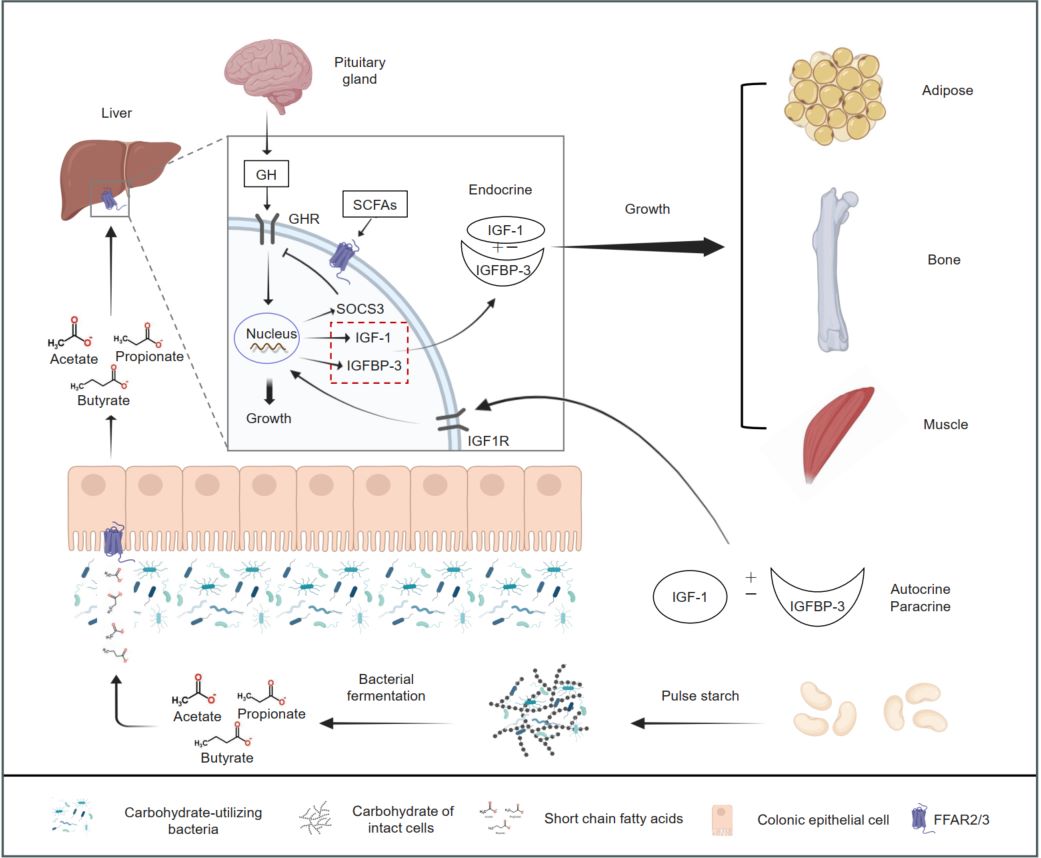
The MAL-IC diet promotes growth in malnourished mice by regulating the gut microbiota—increasing the abundance of carbohydrate-utilizing bacteria and SCFAs production. SCFAs circulate to the liver via the gut–liver axis, where they bind FFAR2 and activate the somatotropic axis, thereby stimulating GHR-mediated GH signaling and increasing IGF-1 and IGFBP-3 production, ultimately promoting systemic growth.
Akkermansia muciniphila enhances chicken resistance to infectious bronchitis virus by boosting GABA synthesis and suppressing the NF-κB inflammatory pathway
- 23 February 2026
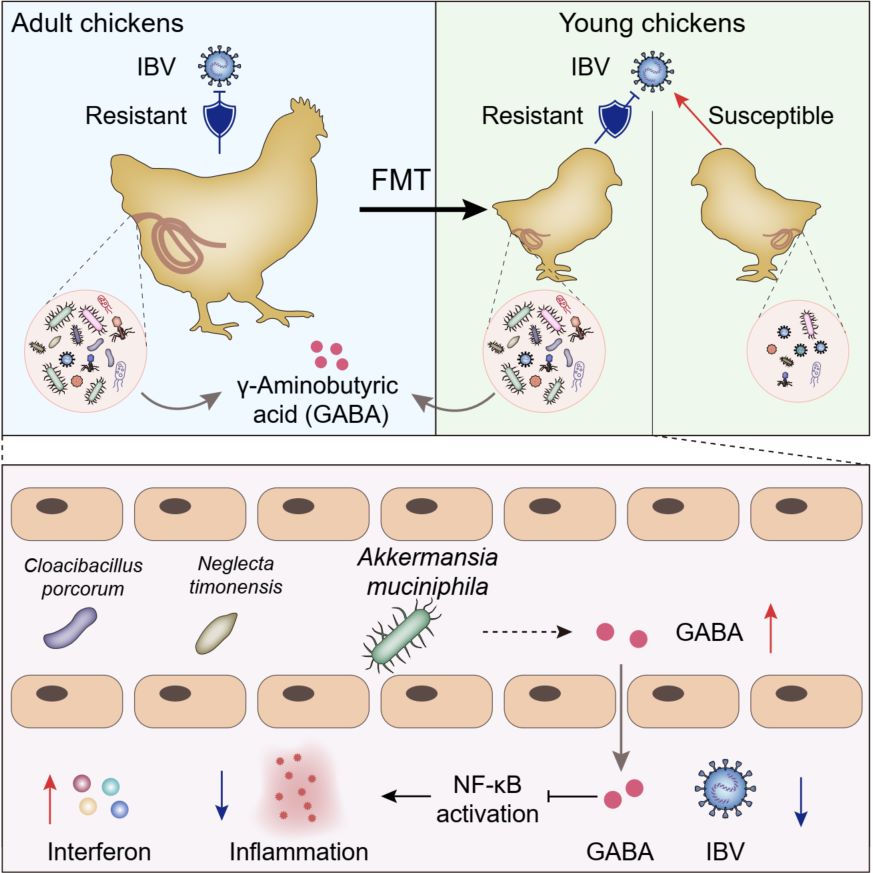
Infectious bronchitis virus (IBV) represents a major threat to poultry production, with current control strategies offering limited protection. In this study, we demonstrate gut microbiome is a critical determinant of IBV resistance using fecal microbiota transplantation. Through an integrated multi-omics approach and a newly developed persistent IBV infection model, we identify Akkermansia muciniphila (A. muciniphila) as a key bacterium that enhances resistance to IBV. Administration of A. muciniphila significantly improves survival rates in young chickens from 30 to 35% to 90–95%. Mechanistic investigations reveal that A. muciniphila enhances γ-aminobutyric acid (GABA) production, which suppresses NF-κB-mediated inflammatory signaling, reduces pro-inflammatory cytokine levels, mitigates nephritis progression, and upregulates antiviral interferon expression. These findings highlight the translational potential of live biotherapeutic products as a novel strategy for IBV control in poultry production.
Move beyond free radical theory of aging
- 04 February 2026
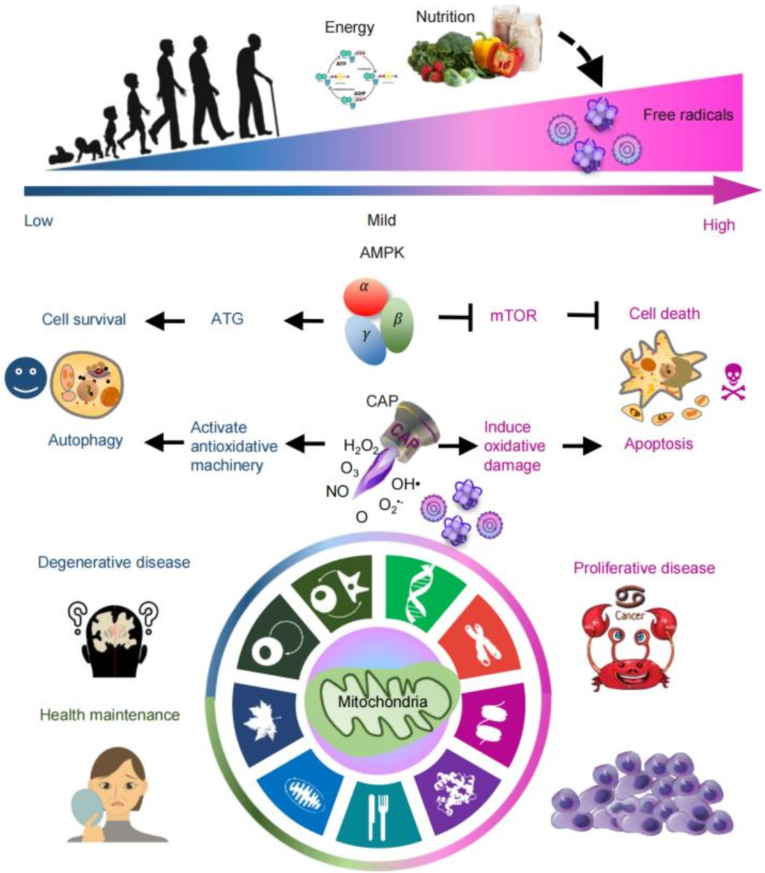
Free radicals play a dual, dose-dependent role in aging. Low levels activate the key sensor adenosine 5'-monophosphate-activated protein kinase (AMPK), promoting cell survival mechanisms like autophagy and enhancing mitochondrial health. High levels, however, push AMPK to initiate cell death pathways, such as apoptosis. This positions AMPK as a central nexus linking free radical signaling to mitochondrial function and aging outcomes. Based on this principle, cold atmospheric plasma (CAP), as a controllable source of reactive species including free radicals, offers therapeutic potential. Lower doses can support health and treat degenerative diseases by boosting cellular resilience and survival. Conversely, higher doses can be applied to eliminate malignant cells, making CAP a promising tool against proliferative diseases like cancers.
Microbiome-urothelium crosstalk in bladder cancer: From dysbiosis to clinical translation
- 14 February 2026
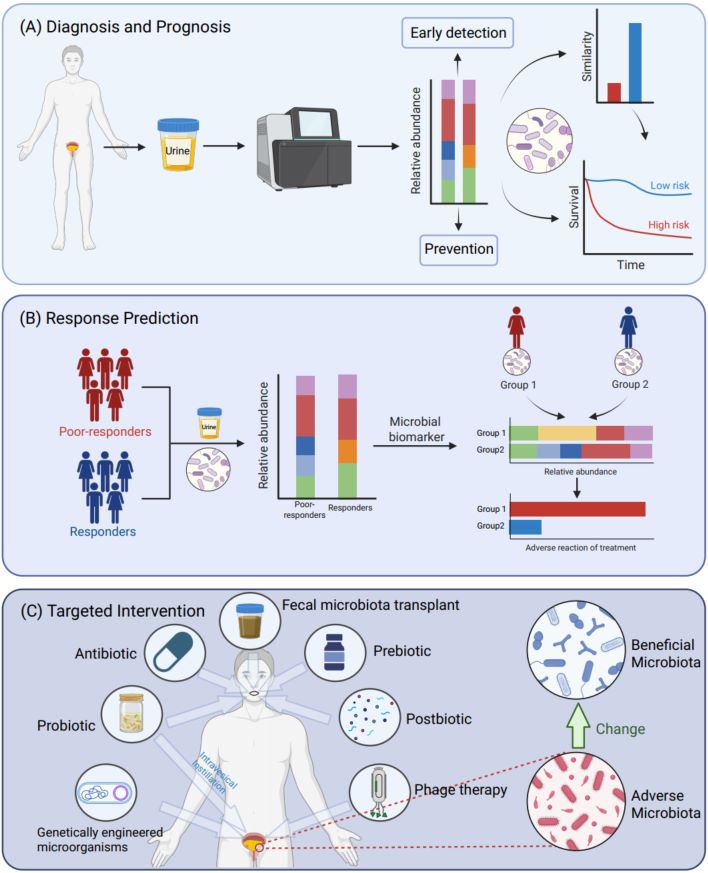
This review elucidates the critical crosstalk between the urobiome and bladder cancer (BCa), mapping the landscape from ecological dysbiosis to clinical translation. We synthesize emerging evidence on microbial signatures that distinguish BCa patients, exploring key carcinogenic mechanisms including chronic inflammation, genotoxicity, and the gut–bladder metabolic axis. Crucially, the review underscores the translational utility of the microbiome as a non-invasive liquid biopsy for risk stratification and for predicting therapeutic responses to Bacillus Calmette Guerin immunotherapy and immune checkpoint inhibitors. Furthermore, we propose a forward-looking framework for precision oncology, integrating microbiome-based interventions—such as probiotics, targeted bacteriophages, and antibiotic stewardship—to modulate the tumor microenvironment and optimize patient outcomes.
Early matrine intervention of gut microbiota for type 2 diabetes prevention
- 05 August 2025
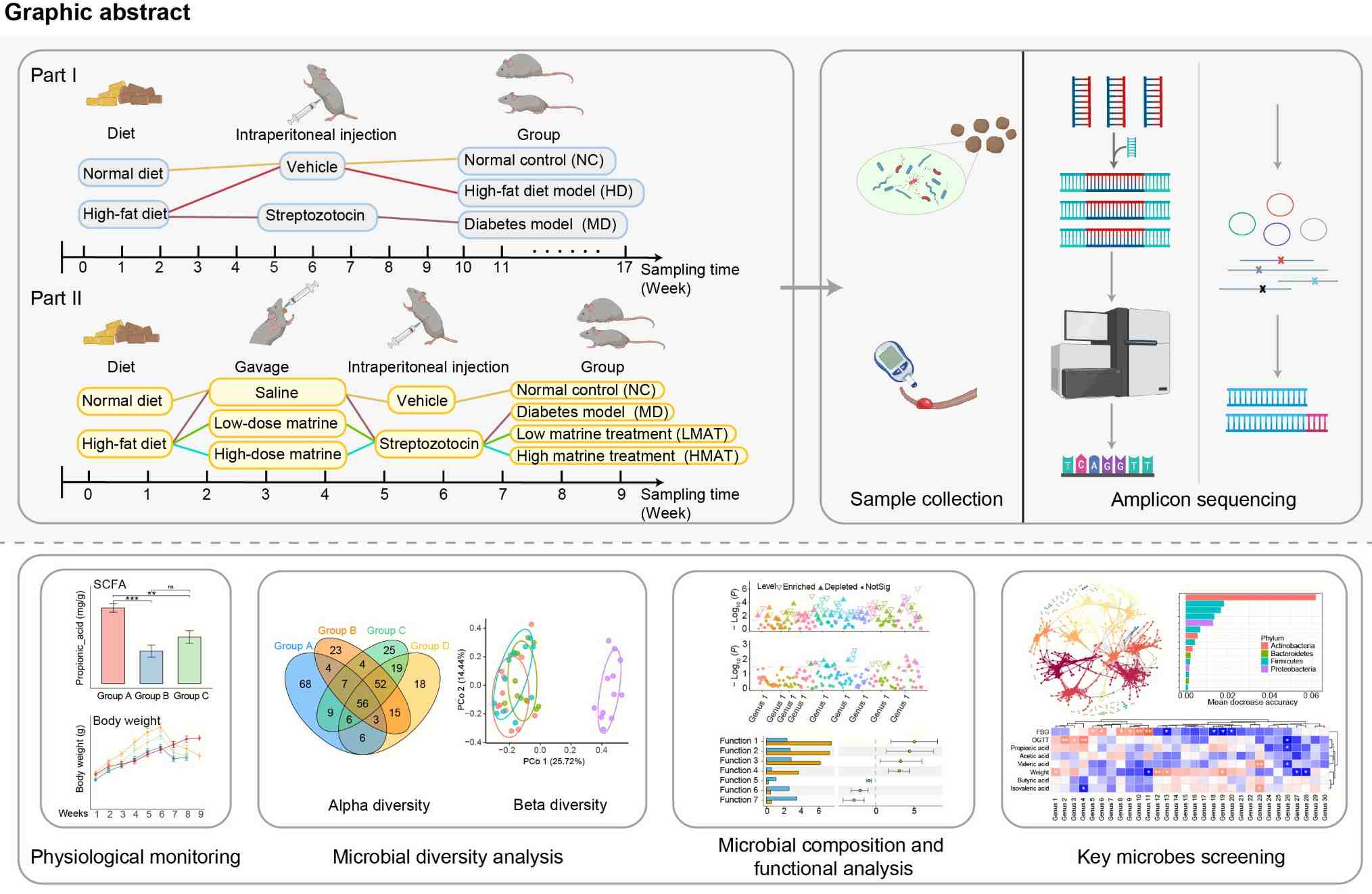
How matrine influences gut microbiota imbalance to prevent the progression of diabetes remains unclear. We conduct experiments using mice to simulate the stages of diabetes development and matrine intervention. Combined with amplicon sequencing, we find that the gut microbiota of diabetic mice continuously changes with the progression of the disease. Furthermore, differences in microbial community composition and function can distinguish the matrine intervention group from other groups. Muribaculum is generally enriched in the intervention group, while Lachnospiraceae_incertae_sedis is predominantly enriched in the diabetes group. These microbes may become new targets for diabetes intervention, providing new insights into diabetes treatment.
Host–microbiota interaction drives 5-hydroxyindole-3-acetic acid production to promote linear growth in infant mice
- 23 October 2025
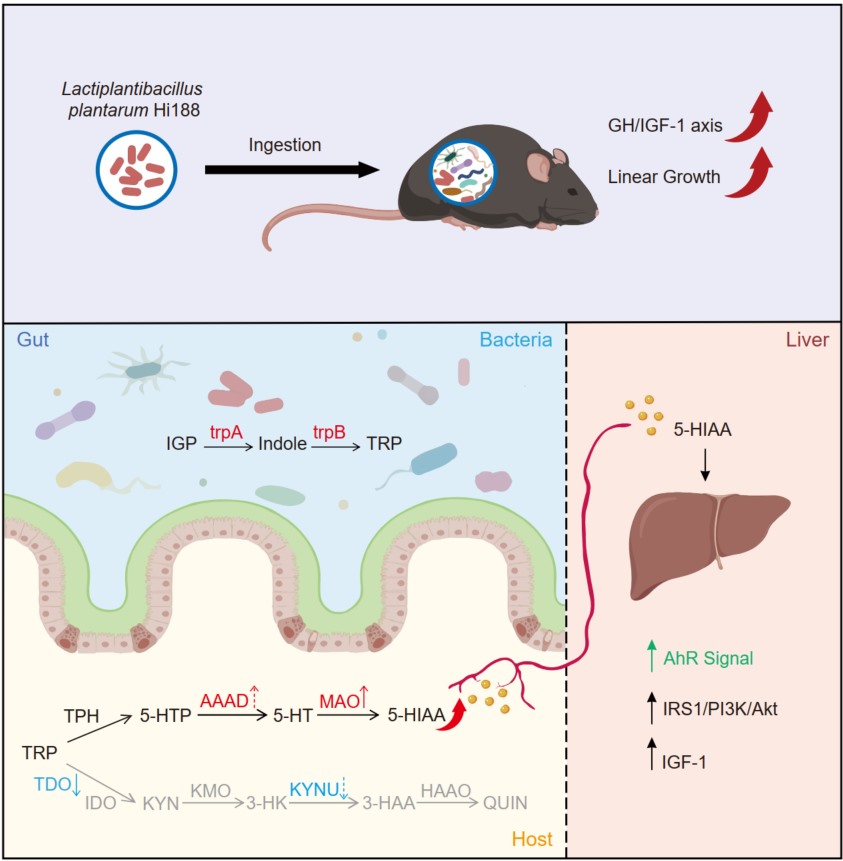
This study discovered that Lactiplantibacillus plantarum Hi188 promoted linear growth in postweaning mice. Transcriptomic analysis, untargeted metabolomics, and in vitro experiments showed that the elevated levels of 5-hydroxyindole-3-acetic acid (5-HIAA) activated the hepatic aryl hydrocarbon receptor (AhR) and subsequently promoted insulin-like growth factor-1 (IGF-1) production via the IRS1/PI3K/Akt pathway. Integrated metagenomics and reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis of host colonic genes revealed that 5-HIAA was cooperatively produced by the gut microbiota and the host. These findings offered novel perspectives on the therapeutic potential of microbiota-targeted interventions for growth regulation.
Ribosomal RNA operon copy number: A trait-informed framework to close the microbial cultivation gap
- 25 December 2025
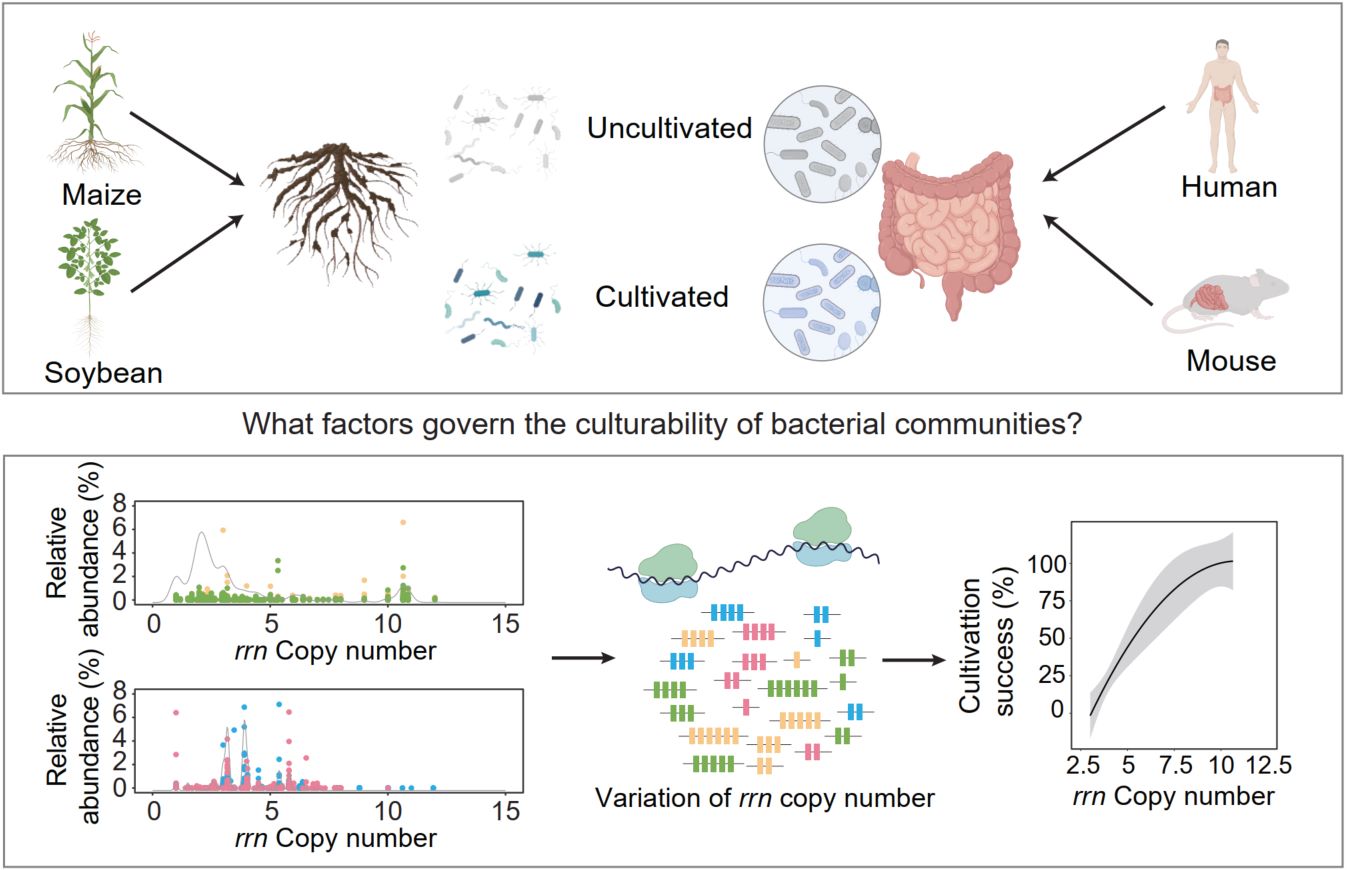
The vast majority of microbes remain uncultured, constraining functional validation and limiting microbiome-based applications. We show that ribosomal RNA operon (rrn) copy number—a stable, phylogenetically conserved genomic trait—is strongly correlated with cultivation outcomes across ecosystems. Comparative analyses of rhizosphere, human gut, and mouse gut microbiomes reveal that rhizosphere communities contain a higher proportion of high-rrn taxa, which are preferentially recovered under nutrient-rich, short-term cultivation, whereas low-rrn taxa are systematically underrepresented. By linking rrn distributions to cultivation success, we propose trait-informed strategies for media design, incubation regimes, co-culture approaches, and preservation methods. This framework provides a predictive, generalizable path to bridge sequencing and cultivation, facilitating biodiversity conservation, sustainable agriculture, and microbiome-based therapeutics.
Metagenomic mining reveals novel Cas12 subtypes and their evolutionary diversification
- 07 October 2025
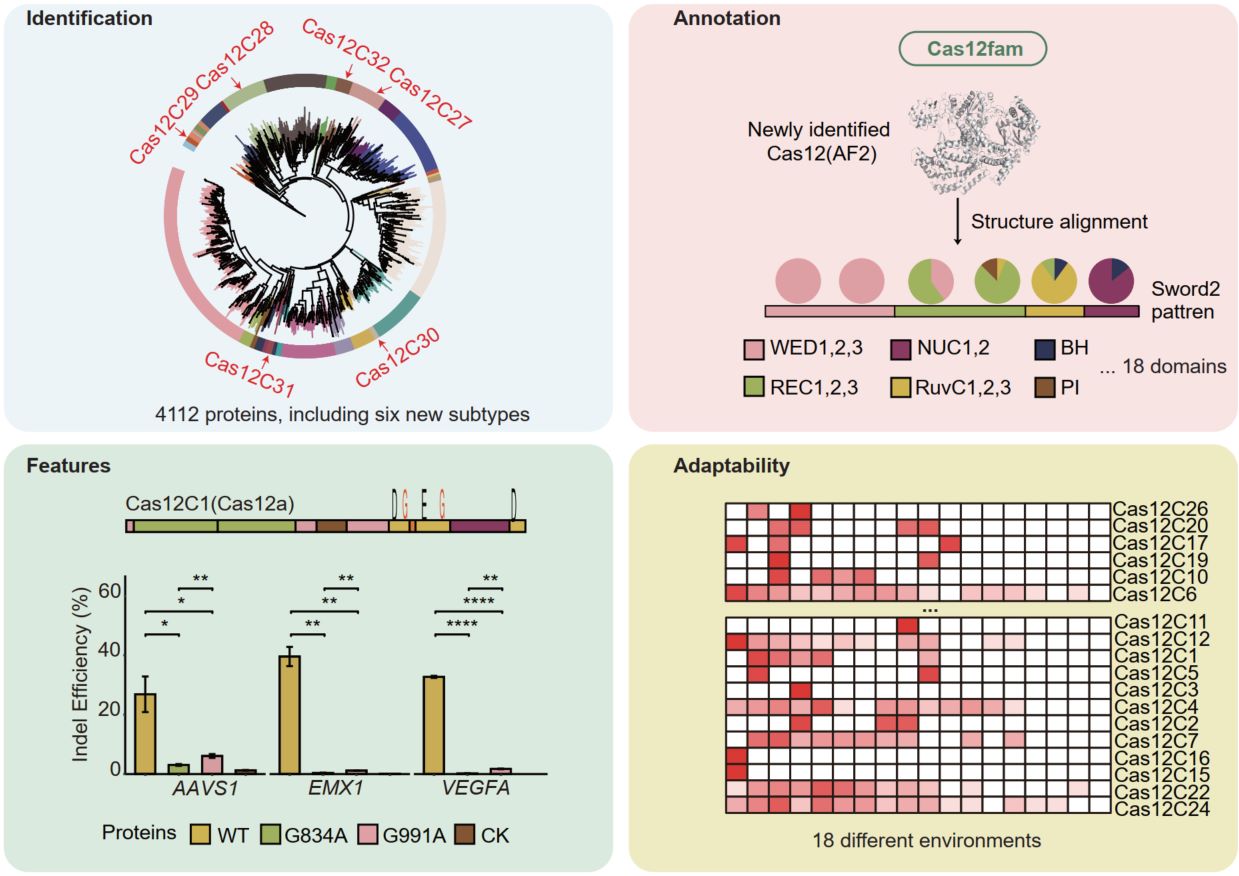
We identified 4112 Cas12 Proteins and 6 new Cas12 subtypes, revealed their significant diversity in N-terminal regions, repeat sequences, and sequences motifs. We developed an AI-driven algorithm, Cas12fam, which allows precise annotation of 18 distinct domains in Cas12 proteins. Through detailed structural analysis, we identified two novel glycine residues in the RuvC-like nuclease domains (RuvC-I, RuvC-II), which were experimentally validated to play a critical role in the DNA-cleavage activity of Cas12. Our analyses revealed that Cas12 proteins are enriched in specific environments, with significant variation in their abundance across ecological niches.
Convergent evolution of metabolic functions: Evidence from the gut microbiomes of humans and dogs
- 21 October 2025
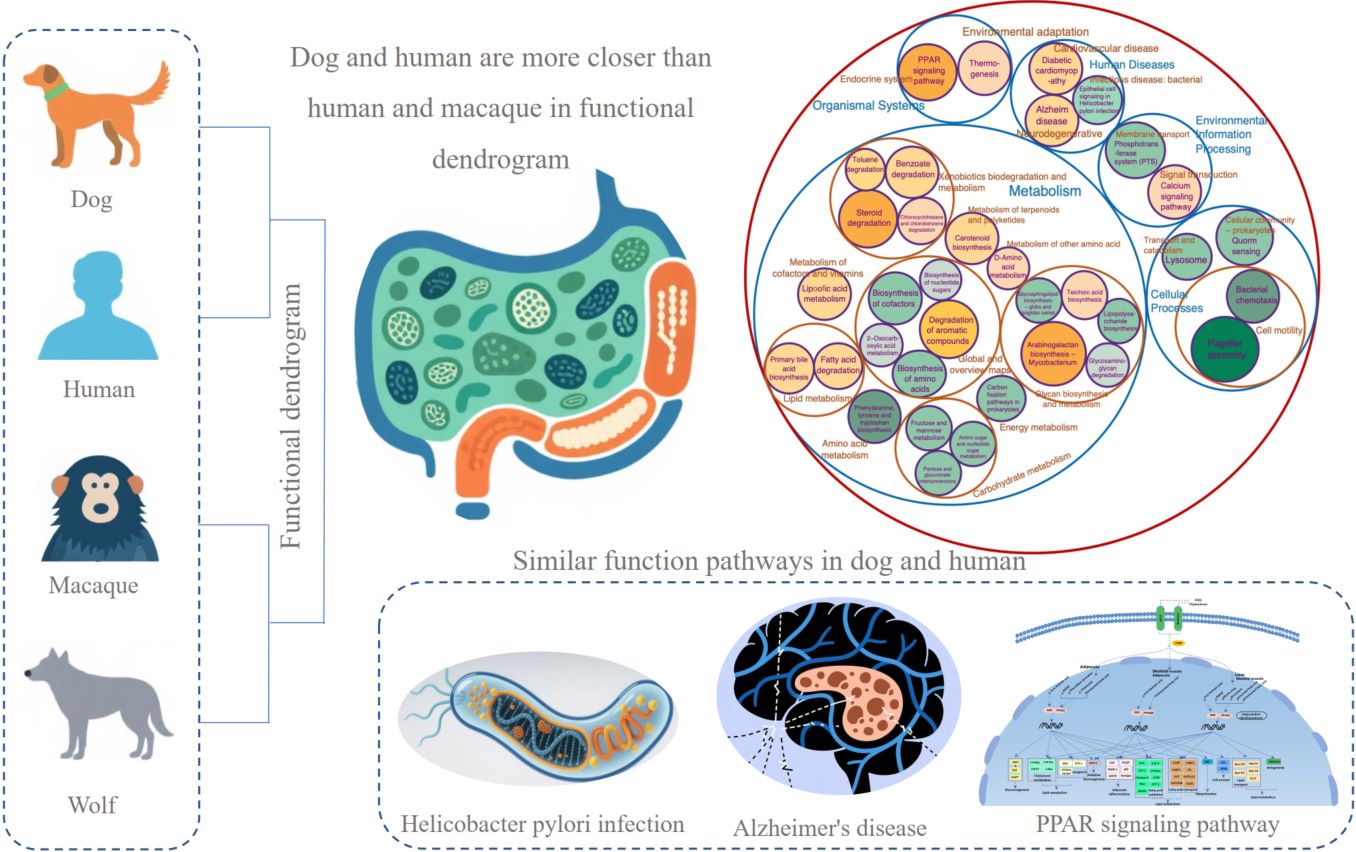
Dogs have undergone convergent evolution in their genome with humans in long-term co-living. Studies have shown that the gut microbiomes of dogs exhibit greater similarity to those of humans compared to mice and pigs. In our study, we first compared the gut microbiomes of macaques, three dog groups, and two wolf groups to those in humans. Results showed that gut microbiomes in dogs have undergone convergent evolution with those in humans. Functional enrichment analysis revealed that dogs exhibit the strongest carbohydrate metabolism ability compared to other hosts including starch and sucrose metabolism. Working dogs and pet dogs showed distinct functional specialization in metabolism and human disease. We suggest that dogs could serve as a potential model for research related to human gut microbial responses to human health, disease, and environmental changes.
Rapid reconstruction of multidrug-resistant bacterial genomes using CycloneSEQ nanopore sequencing
- 26 October 2025
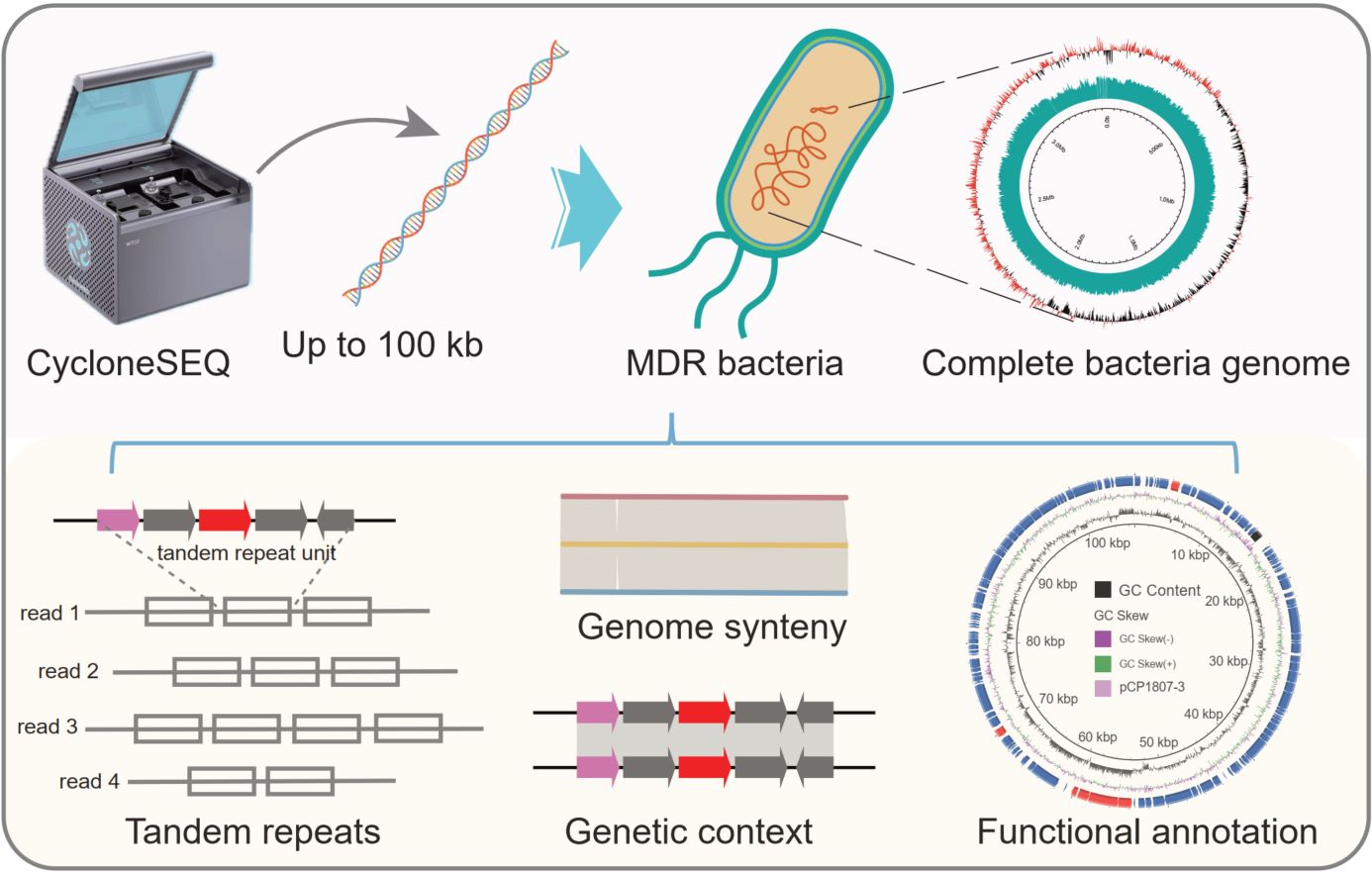
Multidrug-resistant (MDR) bacteria commonly harbor highly complex genomes enriched with mobile elements and resistance islands, which are challenging to resolve using short-read sequencing. Here, we evaluated CycloneSEQ, a newly developed Chinese nanopore sequencing platform, for MDR genome reconstruction. Long-read-only assemblies generated near-complete genomes, while hybrid assemblies achieved accuracy exceeding 99.99%. CycloneSEQ effectively resolved complex structures, including tandem repeats within ARG-bearing regions. Notably, updated sequencing chemistry improved single-read accuracy to 96.2% and yielded assembled genomes with 99.98% accuracy. These results demonstrate that CycloneSEQ enables the generation of complete and highly accurate bacterial genomes, highlighting its potential for antimicrobial resistance research and clinical surveillance.








